







3月9日,马克斯·普朗克生物化学研究所的 Matthias Mann 团队在 Nature Communications 发表了新的研究 AlphaPept: a modern and open framework for MS-based proteomics。
文章介绍了 AlphaPept 这一用于DDA蛋白质质谱数据分析的开源软件。AlphaPept 提供了高效的数据处理、可视化和持续集成功能,其在大规模蛋白质组学研究中具有优越性能和灵活性。

图1 论文截图
AlphaPept框架架构概述
AlphaPept采用了与现有开源项目类似的软件工程设计原则,例如持续测试和集成。通过在GitHub上实施这些原则,使得代码贡献可以通过自动验证的方式进行。这种开放的软件开发模式有助于促进来自不同背景的开发者的贡献和测试,从而维持代码质量。
使用nbdev将文档、代码和测试集成在一个地方,可以自动生成文档,并通过执行笔记本提取生产代码和测试功能。AlphaPept还采用了Numba项目,通过LLVM编译器来直接编译Python算法,实现了代码的高效执行。此外,AlphaPept还可以利用多个CPU核心或GPU对底层算法的计算密集部分进行并行化处理,进一步提高了性能。
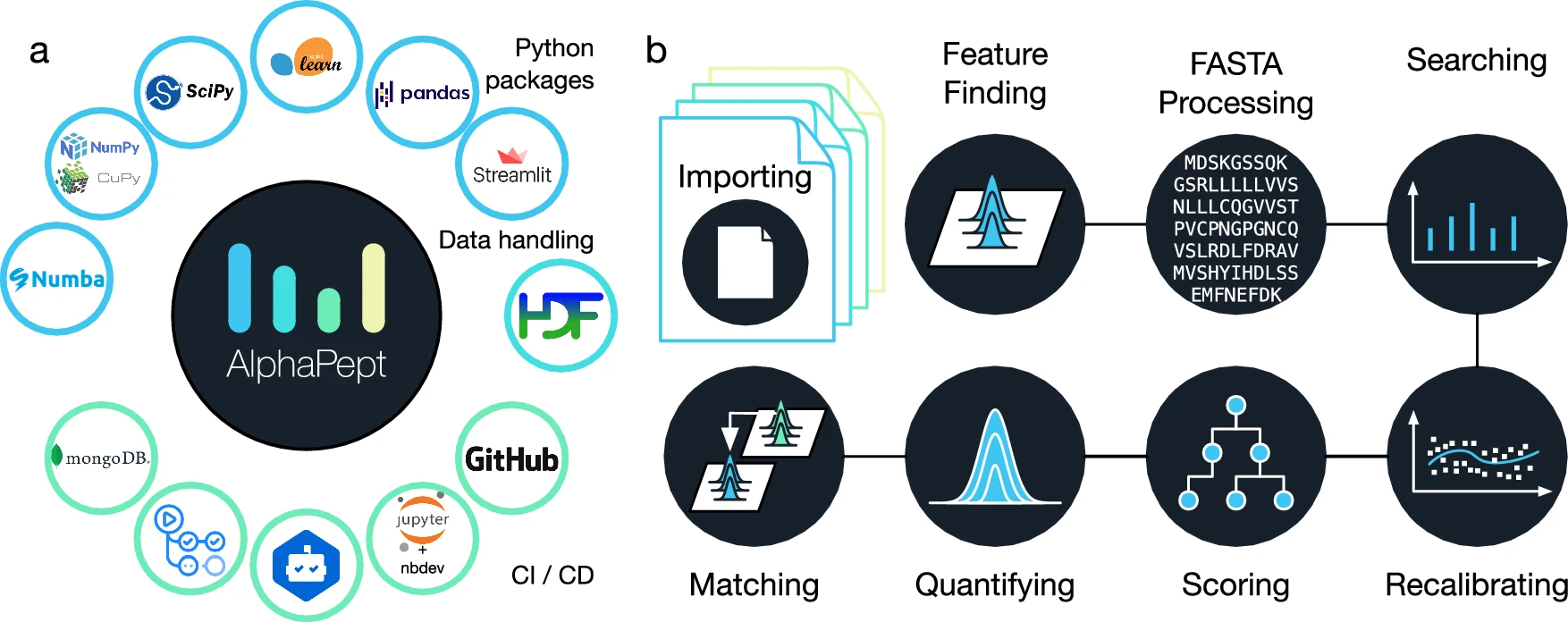
图2 AlphaPept的 “生态系统” 和模块,其依赖于多个经过社区测试的软件包
结果1:
高效且跨平台的质谱数据访问方法
AlphaPept通过多种方式实现了对不同质谱数据格式的高效访问,包括针对Thermo和Bruker的原始数据文件的读取。针对Thermo文件,AlphaPept开发了跨平台的Python API,可以直接读取.RAW格式的质谱数据,并使用了PythonNET来访问Thermo的RawFileReader.NET库。对于Bruker的timsTOF原始数据,AlphaPept也通过Python代码直接处理,借助于Bruker提供的timsdata.dll C/C++库。对于其他供应商的原始数据,AlphaPept则利用Pyteomics库来读取标准的mzML等格式的数据。
AlphaPept选择了HDF5作为存储技术,该格式具有独立于操作系统、任意文件大小、快速访问和灵活的数据结构等优点。同时,AlphaPept 还将中间处理结果存储在 HDF5 容器中,以便可以模块化地执行不同的处理步骤,并且可以从不同的计算机上进行操作。
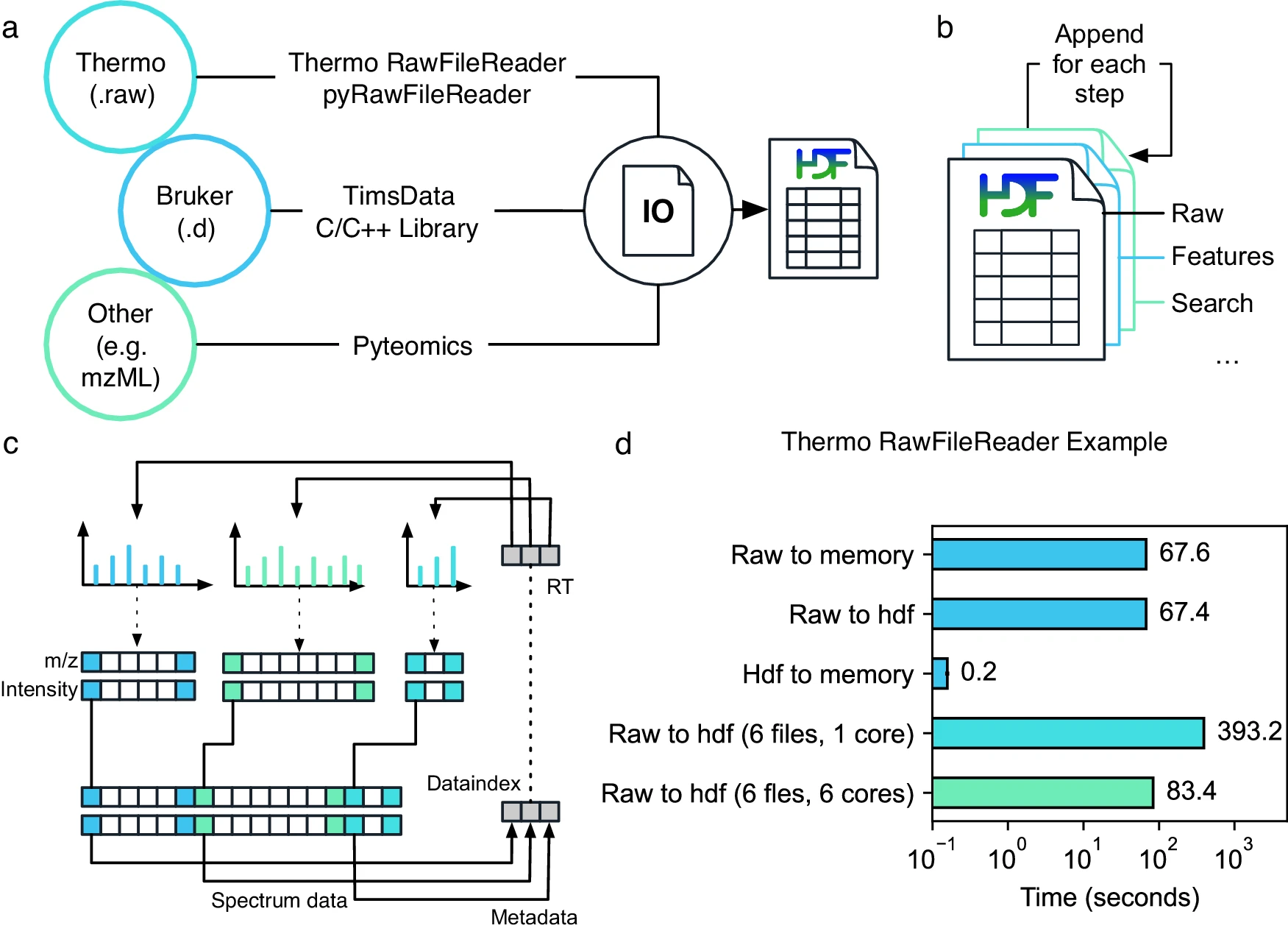
图3 高效且跨平台的质谱数据访问
结果2:
提取母离子同位素峰簇信号(Extracting isotope features)
AlphaPept将质谱峰存储在高效的数据结构中,然后通过计算方法确定色谱洗脱曲线上一级谱图的同位素模式,以支持后续的肽鉴定和定量。采对于Bruker的timsTOF数据,AlphaPept还开发了专门的Python包读取Bruker提供的母离子信号,以实现对离子淌度数据的高效访问和特征提取。
结果3:
肽段-谱图匹配
AlphaPept的核心是将MS2谱图与蛋白质序列数据库中的肽段进行匹配。AlphaPept将蛋白质序列文件解析为肽段,并根据用户指定的规则和氨基酸修饰计算片段质量。通过使用HDF5文件,尽管片段长度不同,但能够有效地存储片段系列。为了实现最大速度,AlphaPept采用了非常快速的碎片计数步骤来确定初始的肽谱匹配。对于人类蛋白组的FASTA文件,通常会生成大约五百万个片段质量的in silico谱图。
结果4:
基于机器学习的评分和FDR估计
AlphaPept使用机器学习算法对肽段-谱图匹配进行评分,以区分真实目标和假目标。该评分模块包括特征提取、候选肽段-谱图匹配子集的选择、机器学习分类器的训练、所有候选肽段-谱图匹配的评分以及通过目标-假目标竞争方法进行FDR估计。通过机器学习算法,AlphaPept显著提高了目标与假目标的区分度,从而获得了更多的肽段-谱图匹配结果。AlphaPept的机器学习评分模块在1% FDR水平上将鉴定率提高了14.4%。
图4 基于机器学习的评分和FDR估计
结果5:
非标记定量
在非标记定量(Label-free quantification)中,需要首先对峰进行积分,然后在样本之间推导出定量比率。AlphaPept对MaxLFQ方法进行了适应,用于非标记定量蛋白质组学数据。
通过解决归一化和蛋白质强度建模的二次最小化问题,AlphaPept实现了非标记定量。AlphaPept利用SciPy包中包含的各种高效求解器来解决这些问题,通过多次比较不同求解器的性能,确定了最佳的求解器,并在实验中获得了验证。
结果6:
MBR和数据集对齐
AlphaPept实现了将MS1特征的标识转移到其他运行的未标识MS1特征的功能(Match-between-runs,MBR)。首先,研究人员通过对保留时间、质量和适用时的离子迁移率进行全局偏移来将多个数据集对齐。然后,将多个运行的前体分组,确定它们的预期特性以及其概率密度,并创建前体库。最后,通过计算每个标识转移的马氏距离,并将其用作概率估计来评估匹配的正确性。
结果7:
使用AlphaPept处理200个HeLa蛋白组
研究人员重新分析了来自最近一项长期性能测试的200个HeLa蛋白组数据集,以展示AlphaPept在大规模研究中的可用性。结果表明,AlphaPept能够与MaxQuant相媲美,识别到约4126个独特的蛋白质组和约47,082个唯一母离子。经LFQ优化后,蛋白质水平的中位数变异系数显著降低。
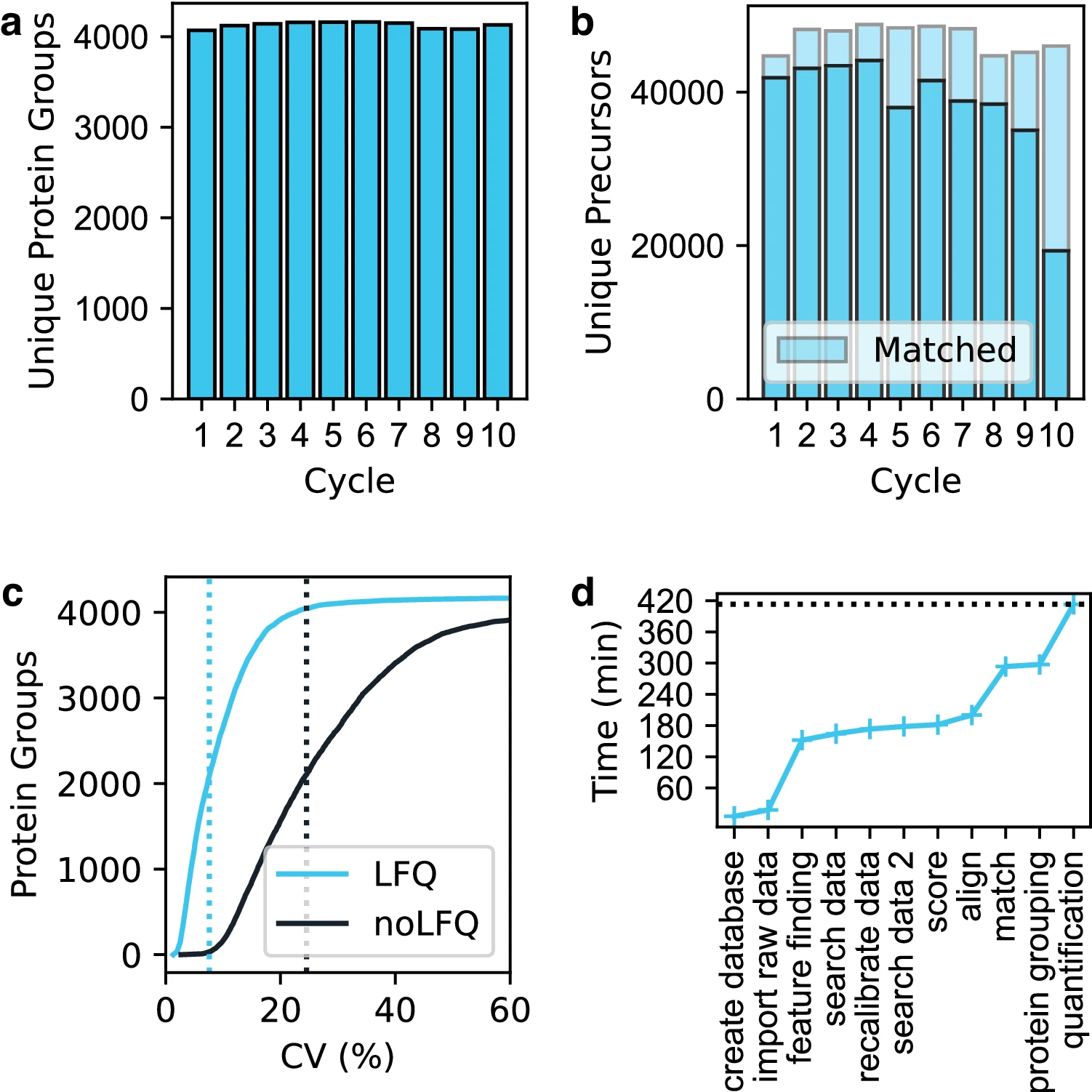
图5 使用AlphaPept处理200个HeLa蛋白组
结果8:
AlphaPept与其他搜索引擎的基准测试
研究人员在一个混合物的LFQ基准数据集上将AlphaPept与MaxQuant和FragPipe进行了基准测试。他们测试了Thermo Orbitrap和Bruker timsTOF条件下的性能,并对比了各个搜索引擎的鉴定和定量性能。结果显示,AlphaPept在酵母条件下的性能介于MaxQuant和MSFragger之间,而在大肠杆菌条件下略逊于MSFragger。综合而言,AlphaPept在标准条件下能够产生与其他常用软件相媲美的搜索结果。
结果9:
标准数据集的持续验证
研究人员使用一系列典型的DDA MS工作流程的数据集进行了持续集成,包括标准单次运行,如HeLa质量控制(QC)运行,以及最近发布的研究。每次对主代码库进行更改后,AlphaPept都会自动重新分析这些文件,以进行全面的系统检查。研究人员还自动上传摘要统计信息,如运行时间、蛋白质数量和特征数量,并在仪表板中可视化这些指标。
结果10:
AlphaPept用户界面和服务器
AlphaPept用户界面和服务器使用了基于服务器的技术,通过Web界面实现用户与软件的交互。用户可以通过连接到本地服务器实例来使用AlphaPept,也可以在强大的处理计算机上运行AlphaPept,并从多个设备访问。AlphaPept使用了名为streamlit的Python软件包来构建服务器功能,使得AlphaPept具备了强大且响应迅速的服务器基础设施。
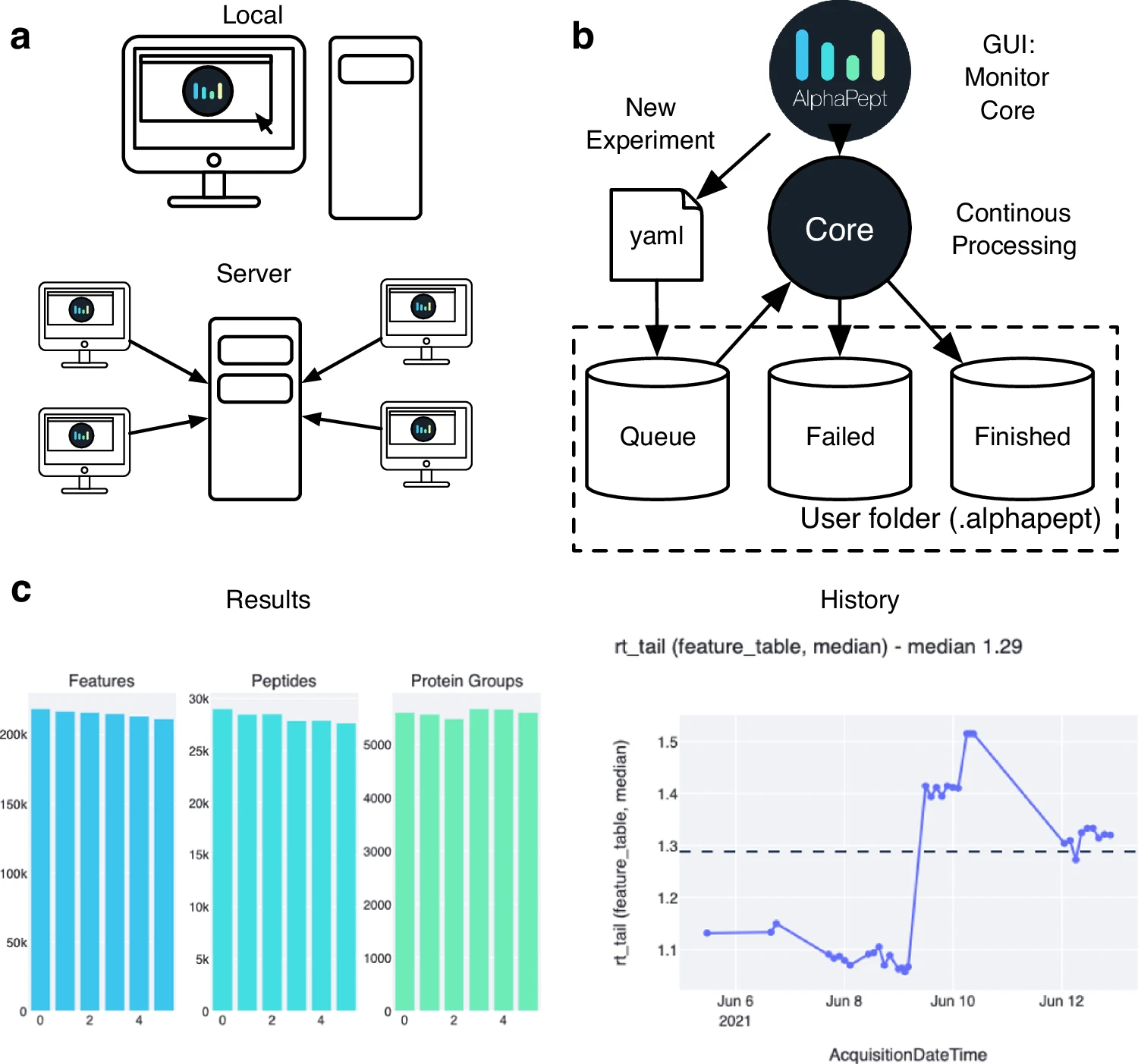
图6 Alphapept 用户界面、工作流程管理、部署和集成
结果11:
AlphaPept工作流管理系统
AlphaPept的workflow管理系统允许用户将处理功能从单个实验扩展到连续处理和监控框架。它使用YAML格式存储实验参数,并在用户文件夹中创建队列、失败和完成文件夹来监视和管理实验。用户可以通过简单检查文件夹来手动检查处理队列,也可以通过编写自定义脚本轻松修改处理队列。AlphaPept还具有文件监视器模块,可自动将新的原始文件添加到处理队列中。
结果12:
结果可视化和持续处理
通过使用streamlit应用程序,可以轻松访问之前处理文件的摘要信息,并在“Finished”文件夹中存储这些信息。AlphaPept还提供了一个History tab,,汇总了以前的结果,以显示随时间的性能变化或分析的MS运行情况。用户可以选择绘制各种摘要统计信息,如已识别的蛋白质或肽段,以及色谱信息(如峰宽或峰尾)。
此外,AlphaPept的浏览器界面还可以绘制实验的识别和定量摘要信息,提供基本的数据分析功能,如火山图、散点图和主成分分析(PCA)。
结果13:
AlphaPept的部署和集成
AlphaPept的部署和集成方式具有多样性。首先,提供了一键安装程序,适用于标准的Windows系统,无需额外的安装步骤。其次,AlphaPept可以作为Python模块使用,扩展了兼容性到其他操作系统。最后,Python模块使得可以将个别功能集成到任何Python程序中,从而创建完全定制的工作流程。AlphaPept还支持在典型的蛋白质组学实验室中连续运行,以自动处理所有新文件,并与Google Colab等云平台兼容。
总 结
在文章讨论部分,研究人员进一步强调了AlphaPept的优点,包括其采用纯Python编写算法、与Python科学生态系统工具的无缝集成,以及其吸引社区参与和贡献的能力等。研究人员还提到了AlphaPept的未来发展方向,包括支持DIA数据分析、更快的数据访问和可视化、深度学习应用、搜索引擎结果的可视化、结果的视觉注释以及统计下游分析的改进。
总的来说,AlphaPept框架旨在提供一个开放、高效、易用的解决方案,以推动质谱学领域的进步,并促进社区合作和贡献。
AlphaPept 完全开源,可在Apache许可证下使用:
https://github.com/MannLabs/alphapept
文章链接:
https://www.nature.com/articles/s41467-024-46485-4


