欧米行业动态
1 2024 HUPO ECR论文竞赛正式拉开帷幕
文献目录
2 OpenMS 3:实现大规模质谱数据的可重复分析
3 Review:“泛组学”+三维体外肿瘤模型,助力药物发现与精准医疗
4 基孔肯雅病毒如何影响大脑和身体?
5 AlphaPept:基于质谱的蛋白质组学开源框架
6 SPLAT:蛋白质组定位和周转分析揭示稳态破坏的时空特征
7 腹水细胞外囊泡蛋白质组学分析揭示卵巢癌肿瘤微环境
8 scmFormer:集成大规模单细胞蛋白质组和转录组学数据
9 帕金森病患者脑脊液、血浆和尿液的蛋白质组学分析
一起来看看本周蛋白组学领域行业动态和精选优质文献吧!
1. 2024 HUPO ECR论文竞赛正式拉开帷幕

2024年HUPO大会将于2024年10月20-24日在德国德雷斯顿(Dresden)举行。现已开放网上注册及摘要提交(包括travel award)通道。
此外,第十届HUPO ECR论文竞赛也已拉开帷幕。博士后研究员、年轻临床医生和初级教职成员可以通过这个独特的平台在蛋白质组学界初露锋芒,展示自己对蛋白质组领域的重要贡献。
竞赛的三名入围者在HUPO 2024大会的专设全体会议上报告于2023年和2024年间发表的论文,届时将由专家委员会评估口头报告,选出 “年度蛋白质组学亮点” (Proteomics Highlight of the Year)。一等奖获得者将获得1000美元的现金奖励,而其他两名入围者将各自获得500美元的奖励。
竞赛将在4月1日截止申请,关于参赛资格、论文提交等更多比赛信息请参见官网通知:
https://www.hupo.org/ECR-Manuscript-Competition
2.(Nat Methods,IF: 48)OpenMS 3:实现大规模质谱数据的可重复分析

上月,德国图宾根大学(University of Tuebingen)Timo Sachsenberg 团队在 Nature Methods 发表了通讯文章:OpenMS 3 enables reproducible analysis of large-scale mass spectrometry data。
文章总结了开源质谱数据分析平台OpenMS 3的新功能和变化,该版本将质谱数据处理扩展到了多个生命科学领域,包括蛋白质组学、代谢组学、结构生物学和寡核苷酸质谱学,并通过现代化的Python接口和改进的文档使计算方法使其更易于使用。
https://www.nature.com/articles/s41592-024-02197-7
3.(Mol Cancer,IF:37.3)Review:“泛组学”+三维体外肿瘤模型,助力药物发现与精准医疗

随着基因组学、转录组学、蛋白质组学、代谢组学和脂质组学等 “泛组学”(pan-omics)技术的出现,科学家们不断加深着包括癌症在内的各种疾病的分子和代谢组学的理解。
此外,三维(3-D)疾病模型已被有效利用于了解疾病的病理生理学以及作为药物发现中的筛选工具。利用泛组学技术和三维体外肿瘤模型的综合方法,已经改进了对实体肿瘤中各种信号通路和分子交互作用的复杂网络的理解。
文中指出,三维疾病模型与蛋白质组学的融合具有巨大的未开发潜力,有望改善药物发现。由于蛋白质组是细胞的主要功能组成部分,并决定它们如何相互 “沟通”,因此在疾病的发生发展中起着重要作用。然而,还需要用蛋白质组学技术(例如基于质谱流式细胞术的单细胞蛋白质组学和空间蛋白质组学)进行进一步研究,以充分探索蛋白质组学和三维疾病模型在药物发现中的潜力。
在综述中,研究人员强调了组学技术的当前趋势,并强调了它们在了解癌症基因型-表型相关性方面与三维体外肿瘤模型的作用。研究人员进一步讨论了与组学技术相关的挑战,并对这些技术在药物发现和精准医学中的未来应用提供了展望。
https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-023-01916-6
4.(Cell Host Microbe,IF:30.3)基孔肯雅病毒如何影响大脑和身体?

基孔肯雅病毒(CHIKV)是一种蚊媒甲病毒,可引起急性、亚急性和慢性的人类关节炎疾病,在极少数情况下也会导致神经并发症和死亡。研究人员结合流行病学、病毒学、组织病理学、细胞因子、分子动力学、代谢组学、蛋白质组学和基因组学分析,研究了导致基孔肯雅病毒相关(CHIK)死亡的病毒和宿主因素。
研究人员发现CHIK幸存者组与健康组相比有525种蛋白质差异表达(268种下调,257种上调),蛋白质组学分析结果表明CHIK死亡中出现了凝血和免疫系统途径的失调。
研究结果表明,与幸存者相比,CHIK死亡与多器官感染、中枢神经系统损伤以及促炎细胞因子和趋化因子水平升高相关。CHIK死亡的组织病理学、代谢物和蛋白质组学特征显示了血液动力学紊乱和免疫反应失调。此外,CHIKV感染破坏了血脑屏障的完整性,这表现为通透性增加和紧密连接蛋白表达异常。
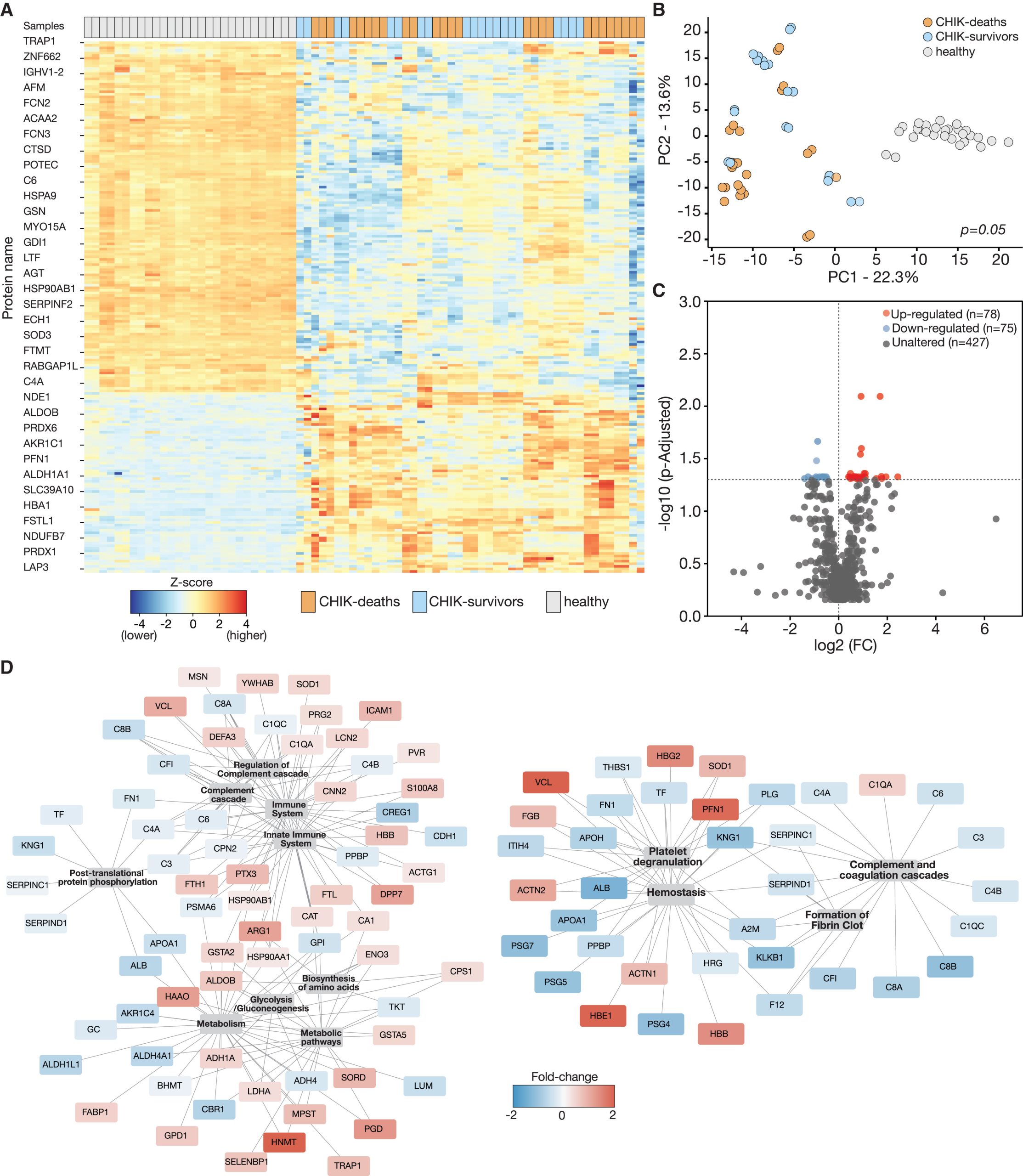
不同疾病结果的CHIK患者的蛋白质组学特征
https://www.cell.com/cell-host-microbe/fulltext/S1931-3128(24)00054-4
5.(Nat Commun,IF: 16.6)AlphaPept:基于质谱的蛋白质组学开源框架

3月9日,马克斯·普朗克生物化学研究所的 Matthias Mann 团队在 Nature Communications 发表了新的研究 AlphaPept: a modern and open framework for MS-based proteomics。
文章介绍了AlphaPept这一用于DDA蛋白质谱数据分析的开源软件。AlphaPept 提供了高效的数据处理、可视化和持续集成功能,其在大规模蛋白质组学研究中具有优越性能和灵活性。
https://www.nature.com/articles/s41467-024-46485-4
6.(Nat Commun,IF: 16.6)SPLAT:蛋白质组定位和周转分析揭示稳态破坏的时空特征

蛋白质的空间和时间分布对于其功能至关重要,但无法通过测量蛋白质丰度来直接评估。研究人员描述了一种基于质谱的蛋白质组学策略——Simultaneous Proteome Localization and Turnover(SPLAT),可在同一实验中同时测量蛋白质周转率和亚细胞定位。
应用该方法,研究人员发现在人AC16细胞中,未折叠蛋白反应(UPR)对蛋白质周转有不同影响,具体取决于它们在亚细胞位置上的不同。内质网和高尔基体中的应激反应蛋白质整体减缓,但在应激反应蛋白质中存在加速现象。与此同时,UPR触发了包括RNA结合蛋白和氨基酸转运蛋白在内的蛋白质的广泛差异定位。
此外,研究人员观察到新合成的蛋白质(包括EGFR)在应激状态下与现有蛋白质库显示不同的定位,类似于蛋白质转运的紊乱。接下来,他们将SPLAT应用于诱导多能干细胞衍生的心肌细胞(iPSC-CM)模型,研究癌症药物卡非佐米治疗后的心肌毒性。然而,卡非佐米对整体平均蛋白质半衰期几乎没有影响,反而可能选择性地破坏肌节蛋白质稳态。
该研究提供了蛋白质空间和时间动态相互作用的视角,并展示了一种检查应激和药物反应中蛋白质稳态调节的方法。
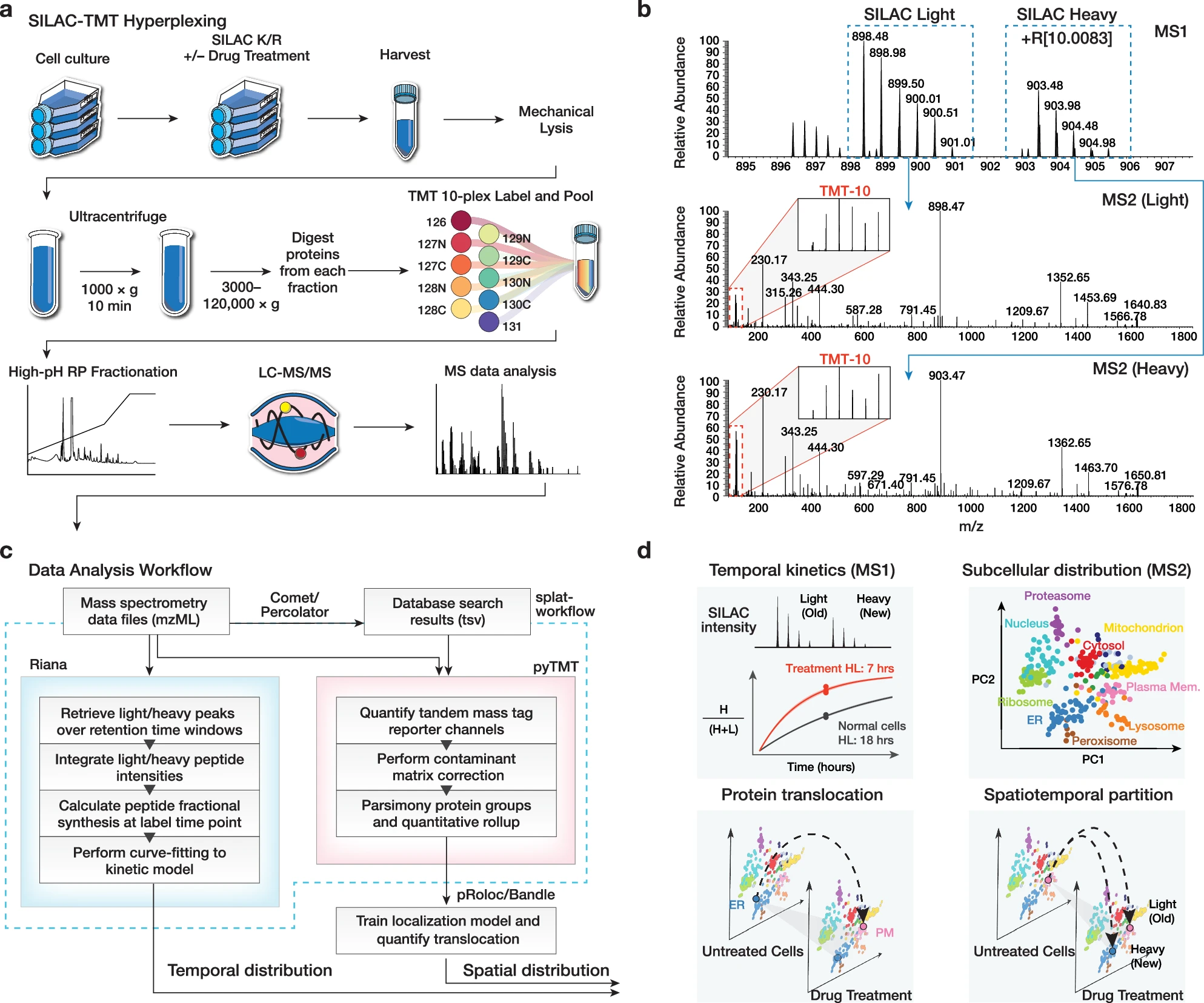
SPLAT策略概述
https://www.nature.com/articles/s41467-024-46600-5?utm_medium=external_display&utm_source=stork&utm_content=email&utm_term=null&utm_campaign=CONR_JRNLS_AWA1_CN_CNPL_0034V_STKRE
7.(J Extracell Vesicles,IF: 16)腹水细胞外囊泡蛋白质组学分析揭示卵巢癌肿瘤微环境

卵巢、输卵管和腹膜的高级别浆液性癌(HGSC)是最常见的卵巢癌类型之一,也是最致命的恶性肿瘤之一。HGSC患者的腹水中包含着各种细胞、蛋白质和细胞外囊泡(EVs)。研究人员通过正交方法从其中分离EVs,并对其进行了蛋白组学分析。
研究人员确定了一组 “核心腹水EV相关蛋白”,并定义了其在HGSC腹水中独特的子集。利用单细胞RNA测序数据,研究人员将HGSC特异性EV的来源映射到存在于腹水中的不同类型的细胞。令人惊讶的是,EVs主要不是来自肿瘤细胞,而是来自非恶性细胞类型(如巨噬细胞和成纤维细胞)。流式细胞术结合匹配样本中的EV蛋白质组成分析显示,在HGSC中分析细胞类型特异性EV标记具有比分析腹水细胞更大的预后潜力。
总之,该研究表明EV的蛋白质组学分析可以定义HGSC肿瘤微环境的细胞组成。这一发现为更好地理解EV在肿瘤促进/预防中的作用和改进HGSC开辟了许多途径。
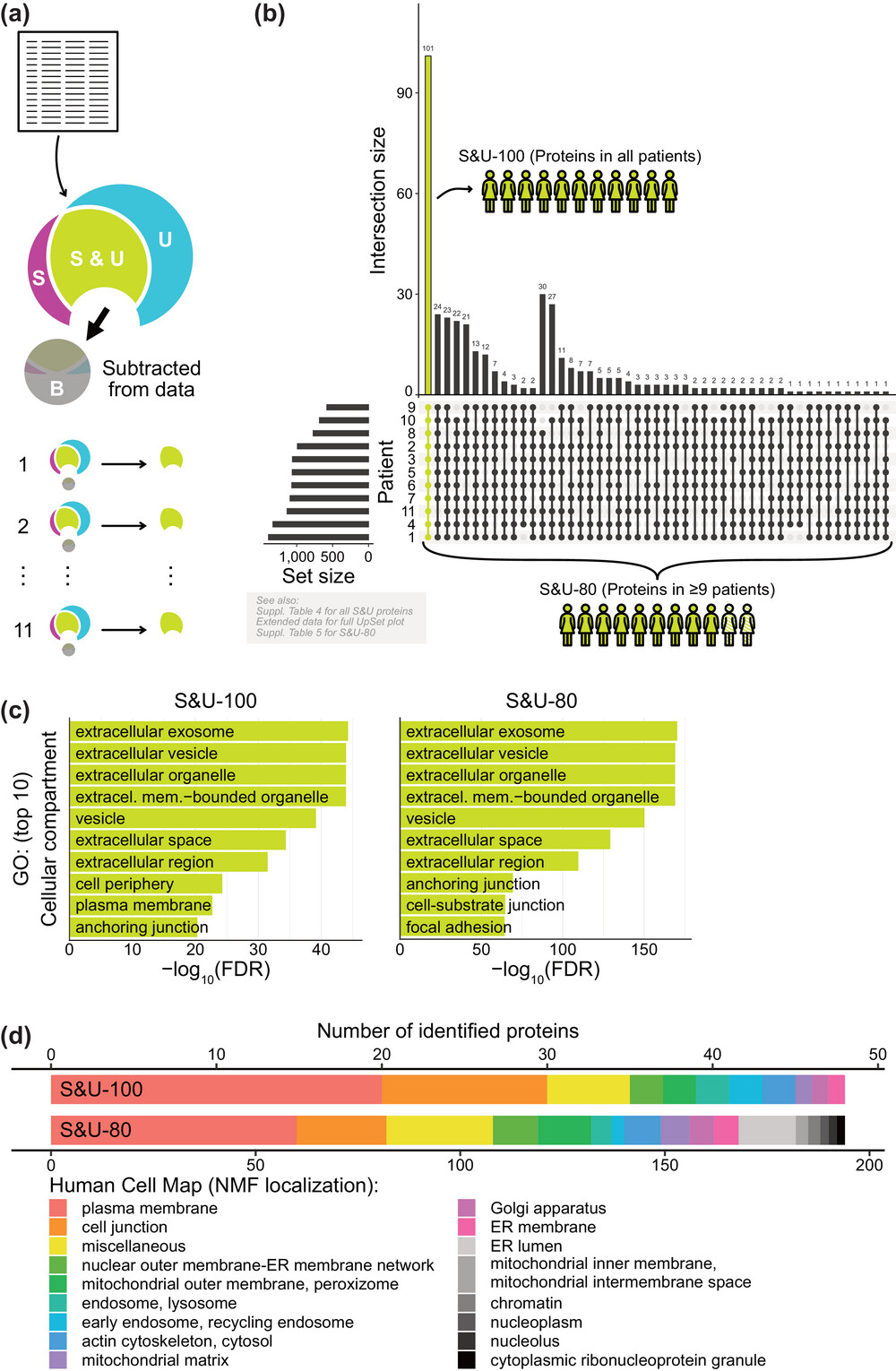
核心腹水EV相关蛋白的鉴定和表征
https://isevjournals.onlinelibrary.wiley.com/doi/10.1002/jev2.12420
8.(Adv Sci (Weinh),IF: 15.8)scmFormer:集成大规模单细胞蛋白质组和转录组学数据

基于Transformer的模型已经彻底改变了单细胞RNA测序(scRNA-seq)数据分析。然而,它们的适用性受到单细胞多组学数据的复杂性和规模的挑战。本文提出了一种新颖的单细胞多模态/多任务转换器(scmFormer),以填补单细胞蛋白质组学与其他组学数据集成的现有空白。
通过系统的基准测试,研究人员证明了scmFormer在整合大规模单细胞多模态数据和异质性多批次配对多组学数据方面表现出色,同时保留了批次间的共享信息和不同的生物学信息。在将细胞类型标签从单细胞转录组学转移到蛋白质组学数据方面,scmFormer的平均F1分数比第二种方法高出 54.5%。使用COVID-19数据集,scmFormer成功地在个人计算机上整合了超过148万个细胞。
此外,研究还证明了scmFormer在生成未测量的模态方面优于现有方法,并且非常适合空间多组学数据。因此,scmFormer是分析单细胞多组学数据的强大而全面的工具。
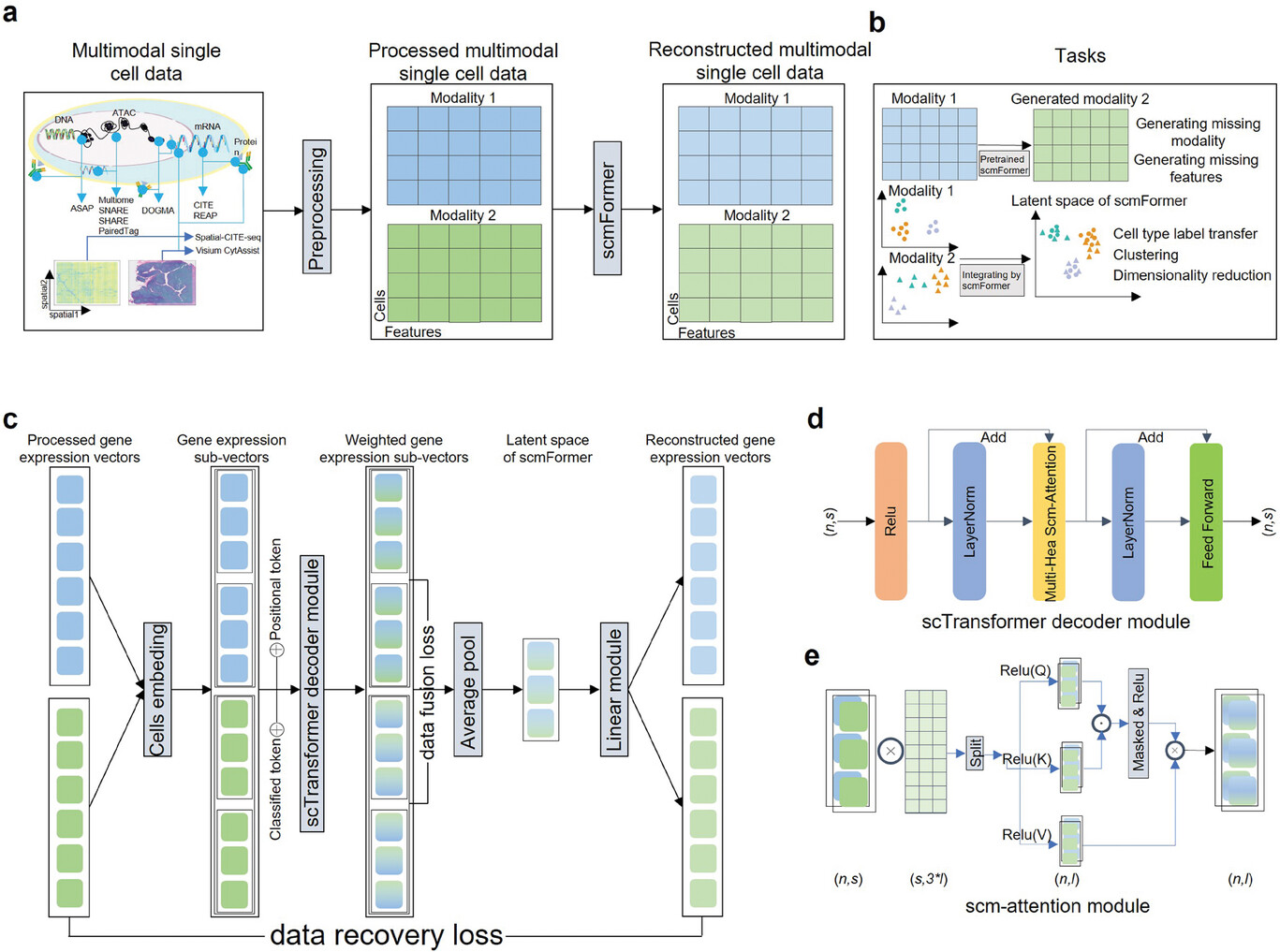
scmFormer概述
https://onlinelibrary.wiley.com/doi/10.1002/advs.202307835
9.(Acta Neuropathol,IF: 12.7)帕金森病患者脑脊液、血浆和尿液的蛋白质组学分析

研究人员进行了一项大规模、多组织、多平台的蛋白质组学研究,以寻找帕金森病早期诊断和疾病监测的新生物标志物。他们使用三种正交蛋白质组学方法对来自七个队列的4877份脑脊液、血浆和尿液样本进行了分析。研究发现数百种蛋白质在帕金森病患者、前驱帕金森病患者(具有DAT缺陷和REM睡眠行为障碍或嗅觉障碍)以及无症状遗传携带者(LRRK2和GBA突变)的脑脊液、血液或尿液中上调。
研究人员在分析中提名了多个新的候选药物作为早期PD的潜力标志物(包括多巴脱羧酶DDC、AADC、SUMF1等)。在多巴胺合成最后一步中催化作用的DDC,作为与PD发病机制具有令人信服的机制联系的新药而脱颖而出。
所有三种蛋白质组学方法都表明,DDC在未接受治疗的PD、前驱PD和GBA或LRRK2携带者的参与者的脑脊液和尿液中保持上调。研究人员发现,脑脊液中的DDC水平与未接受治疗的PD患者的临床症状严重程度相关,并可用于准确诊断PD和前驱PD。这表明尿液和脑脊液中的DDC可能是一种有前景的诊断和预后标志物,在临床护理和转化研究中具有实用价值。
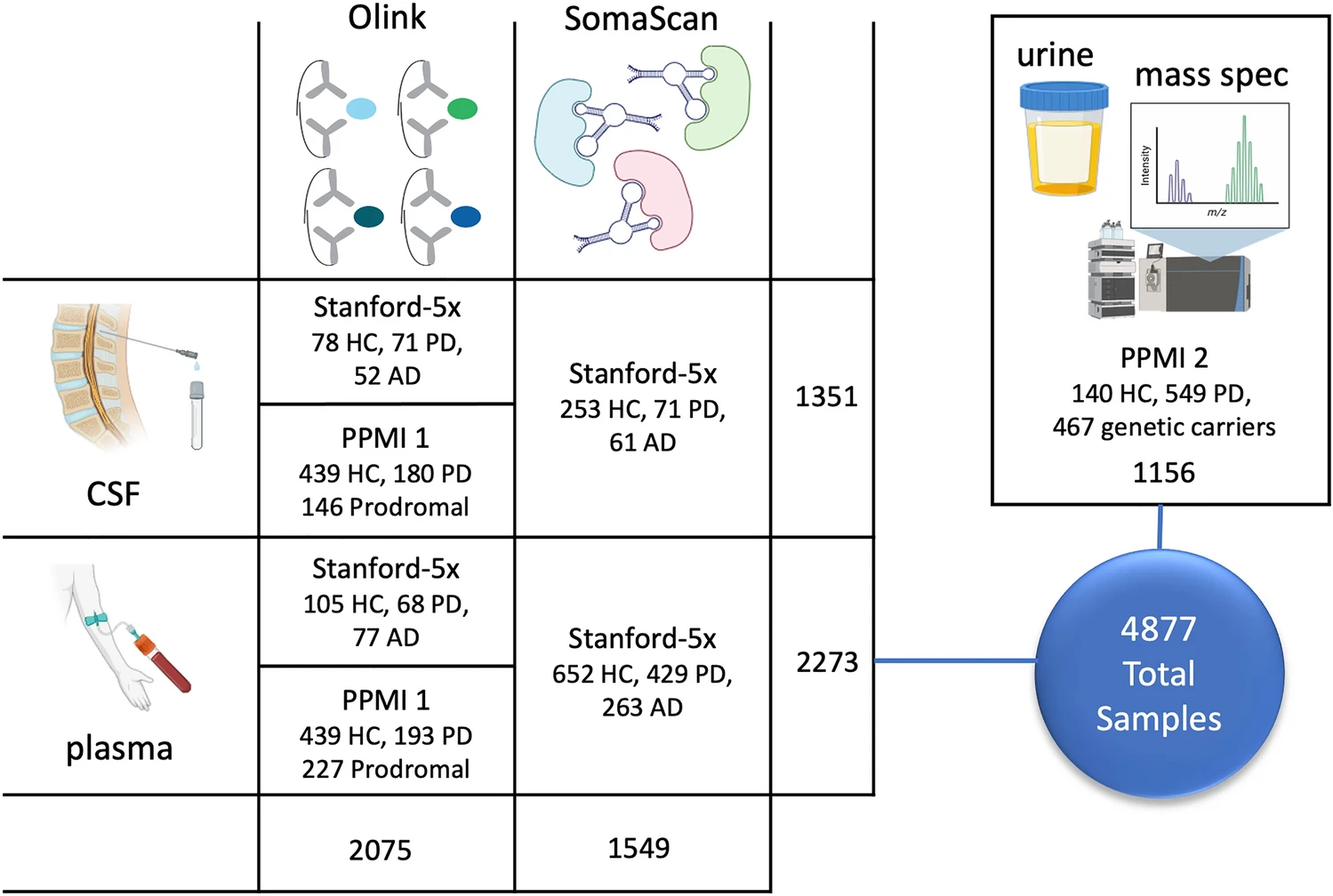
https://link.springer.com/article/10.1007/s00401-024-02706-0?utm_medium=external_display&utm_source=stork&utm_content=email&utm_term=null&utm_campaign=CONR_JRNLS_AWA1_CN_CNPL_0034V_STKRE