乳腺癌是最常见的女性恶性肿瘤,需要有效的治疗策略。分子亚型的出现彻底改变了对这种异质性疾病的理解,使得临床管理更加个体化。尽管已经取得了一定进展,但当前乳腺癌的多组学研究仍存在一些限制,包括样本量不足、临床转化不足以及缺乏亚洲人群数据。
2024年2月12日,来自复旦大学附属肿瘤医院的邵志敏教授、江一舟研究员团队,联合上海市生物医药技术研究院黄薇教授团队,以及复旦大学生命科学学院和人类表型组研究院石乐明教授、郑媛婷副教授团队,在 Nature Cancer(IF=22.7)上发表了题为Integrated multiomic profiling of breast cancer in Chinese population reveals patient stratification and therapeutic vulnerabilities 的研究文章。西湖欧米负责该研究中蛋白质组部分的样本采集、质谱数据分析及生信分析。
基于中国乳腺癌基因组图谱项目(CBCGA),文章研究了中国乳腺癌患者的临床和分子特征,包括肿瘤微环境、基因突变、多组学数据及其相互关系,并利用机器学习模型进行了预测和分类,为了解乳腺癌的病理特征、临床表现和治疗策略提供了重要的线索和数据支持。

图1 论文截图
研究样本
研究纳入了2013-2014年期间在复旦大学附属肿瘤医院接受治疗的来自全国的773名乳腺癌患者(中位随访时间为 83.1 个月),并分析了其基因组学、转录组学、蛋白质组学和代谢组学等多组学数据。
结果1:
CBCGA队列的多组学景观
研究人员对653例样本进行了肿瘤组织和配对血液样本的全外显子测序(WES)分析,对685例样本进行了拷贝数变异(CNA)数据分析,对752例样本进行了RNA测序(RNA-seq)数据分析,对278例样本进行了蛋白质组数据分析,对453例样本进行了代谢组数据分析。此外,还从磁共振成像(MRI)获得了匹配的影像组学数据(n = 419),以及从H&E染色切片获取了数字病理图像(n = 626)。
在整个队列中检测到33,330个非同义突变,其中包括32,037个单核苷酸变异(SNV)和1,293个小插入和缺失。通过PAM50基因预测分析乳腺癌的内在亚型,研究人员发现在752例RNA-seq数据中,29.5%为luminal A型、29.4%为luminal B型、19.7%为HER2富集型、14.9%为基底样型、6.5%为正常样亚型。
此外,研究人员还进行了CNA的评估,并研究了PAM50亚型间差异表达的信使RNA、蛋白质和代谢物。
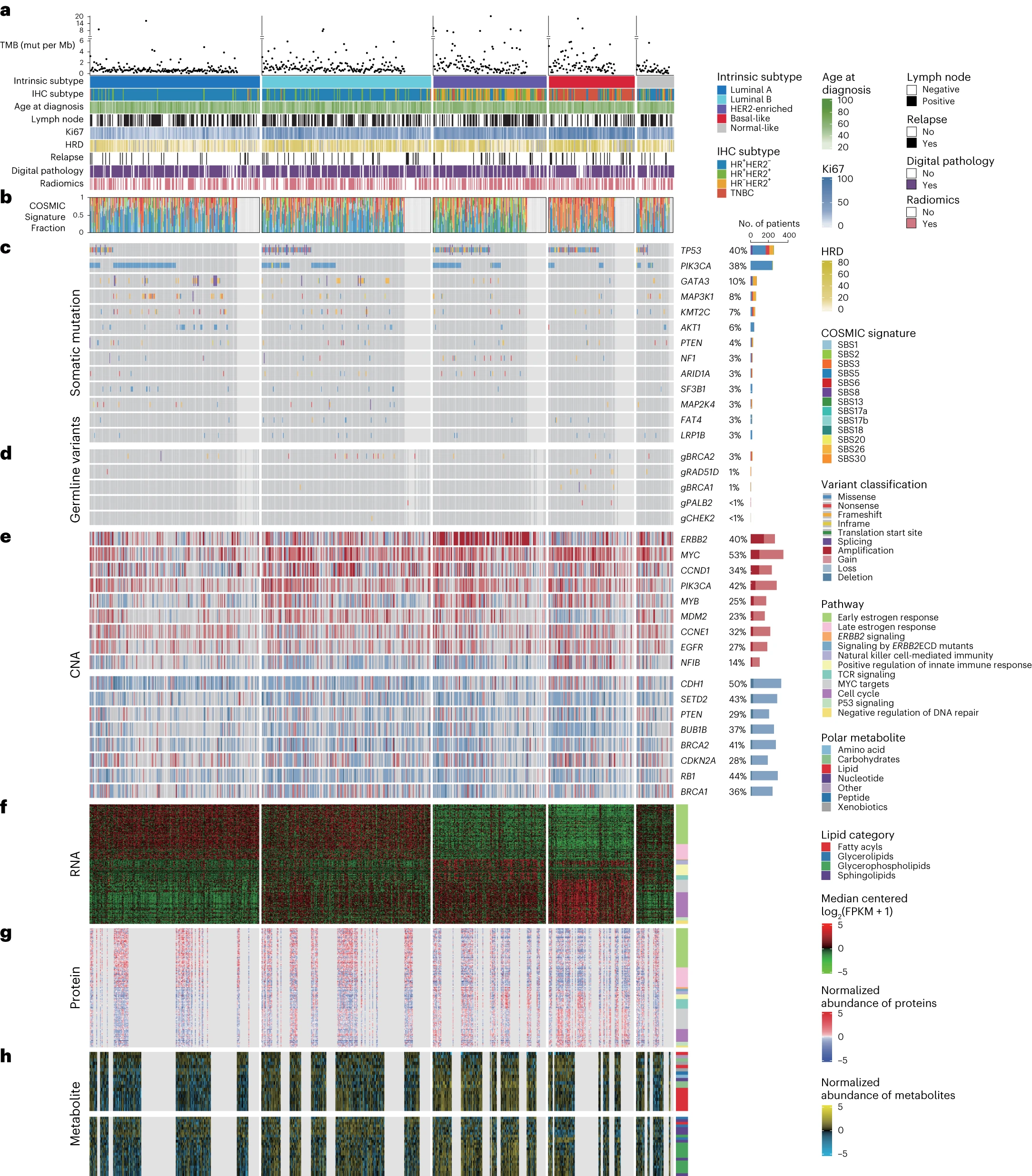
图2 CBCGA队列的多组学景观
结果2:
中国患者乳腺癌的特异性分子特征
研究人员分析了中国患者乳腺癌的临床和分子特征,并在这一背景下探讨了祖源特异性。研究发现,中国患者乳腺癌中AKT1基因的突变频率明显高于TCGA队列(The cancer genome atlas,癌症基因组图谱),尤其在luminal A亚型中更为显著。
同时,中国患者乳腺癌的内在亚型分布与TCGA队列存在差异,HER2富集型亚型在中国队列中比例较高,ERBB2基因扩增更为显著。这些结果提示了中国患者乳腺癌在分子水平上存在特异性,可能需要相应的治疗策略。
结果3:
蛋白质基因组学分析为乳腺癌亚型提供了新的见解
通过使用同位素标记的串联质谱标记(TMT)技术,研究人员鉴定了9787个独特的蛋白质,并研究了拷贝数变异对mRNA和蛋白质水平的影响。结果显示,在不同的内在亚型中,CNA与mRNA和蛋白质水平的相关性不同。他们进一步探讨了潜在的亚型特异性驱动基因和可靶向的CNA事件,发现一些在luminal B亚型中显著的放大频率和CNA-mRNA-蛋白同向效应。
通过综合蛋白质和mRNA丰度,研究人员采用相似性网络融合(SNF)分析进一步细化了乳腺癌患者的分类,发现了一个高风险亚群,该亚群可能受益于免疫治疗。
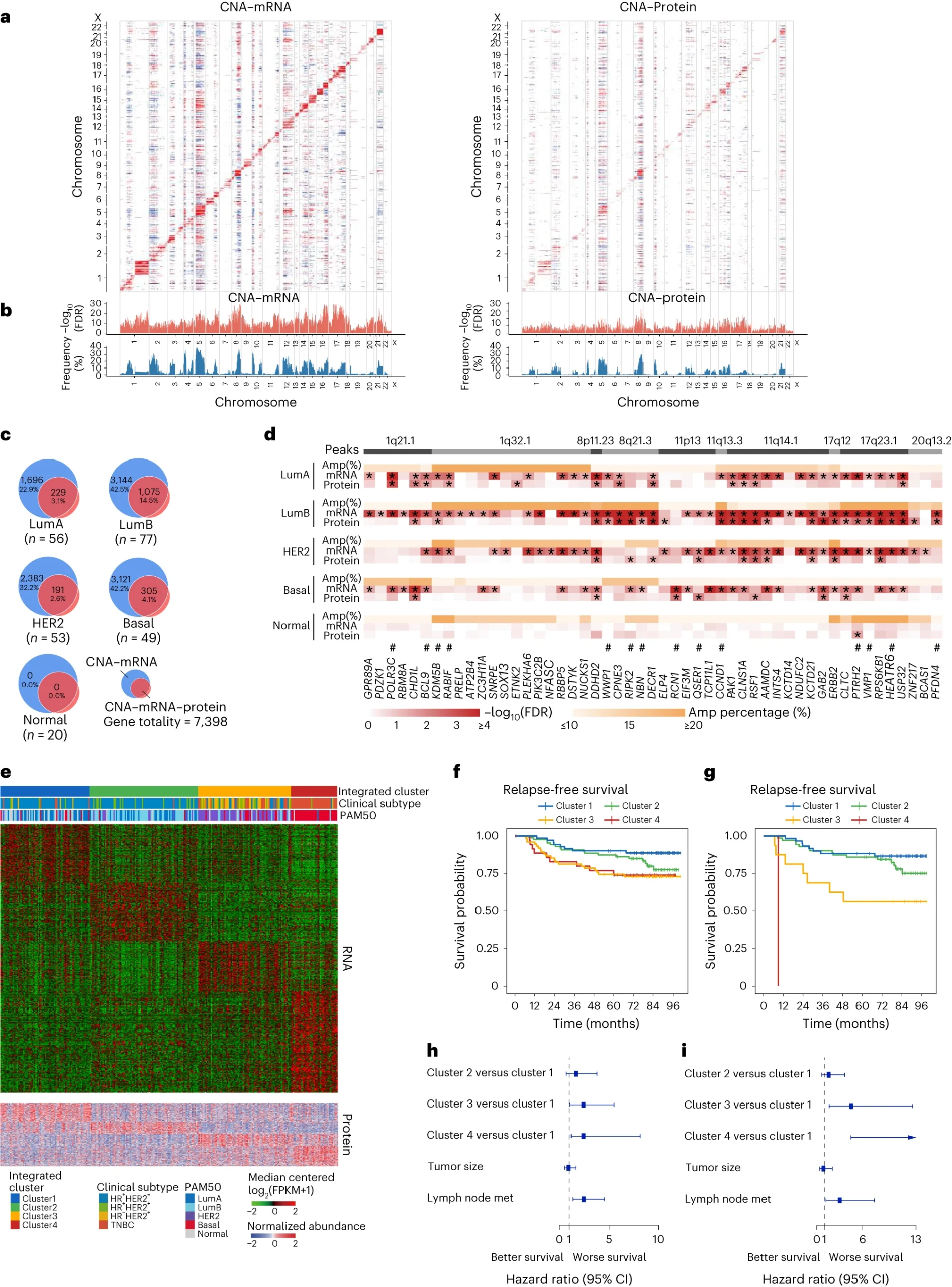
图3 蛋白质基因组学分析为乳腺癌亚型提供了新的见解
结果4:
综合代谢组学分析揭示了铁死亡是基底样肿瘤的潜在治疗靶点
研究人员对CBCGA队列中的669种极性代谢物和1,312种脂质进行了定量分析,结果显示肿瘤和正常样本在极性代谢组学和脂质组学的t-SNE图中明显分离,这表明肿瘤存在明显的代谢失调。
进一步分析发现,与健康组织相比,大多数代谢物在肿瘤中显著富集,特别是氨基酸和肽类显著上调,脂质代谢也异常活跃。通过网络分析识别高度相连的代谢蛋白和代谢物,并发现它们通常也是亚型特异性的,这表明每种乳腺癌亚型具有不同的代谢特征。特别是,研究人员在基底样肿瘤中发现了丰富的铁死亡(ferroptosis)相关物质,这表明铁死亡可能是基底样肿瘤的潜在治疗靶点。
结果5:
免疫基因组学分析揭示了乳腺癌肿瘤微环境异质性
利用乳腺癌的转录组数据,研究人员评估了22种不同的免疫细胞类型和2种基质细胞类型的丰度水平,确定了乳腺癌中三种不同的肿瘤微环境(TME)表型:“冷”(cold TME) 、“中度”(moderate TME)和“热”(hot TME),它们分别代表了低、中和高水平的免疫细胞浸润
免疫基因组学特征的进一步研究揭示了不同TME亚型的T细胞受体多样性和克隆性、肿瘤抗原水平、重要的基因表达和突变特征等差异,为乳腺癌肿瘤微环境的异质性提供了更深入的认识。
结果6:
HER2阳性肿瘤中的复发性ERBB2融合转录本
研究人员利用Arriba和STAR-Fusion分析了CBCGA队列的RNA-seq数据,发现融合基因事件更频繁地出现在17号染色体上,尤其在HER2+样本中。研究人员共检测到11个复发性融合事件,其中一个(LHFPL5–CLPSL1)以前未曾报道过。
进一步的生存分析显示,携带 ERBB2 融合的患者相比于没有携带的患者有更好的生存率,这些数据为了解乳腺癌中的ERBB2融合提供了新的见解,并确定了一些融合作为HER2+乳腺癌的潜在预后标志物。
结果7:
利用机器学习方法整合多模态数据,将乳腺癌患者分为不同的复发风险组别
研究人员从对比增强MRI和病理学切片中获取了影像学和病理学特征,然后结合临床分期(C)、免疫组化亚型、影像学特征、病理学特征(P)、转录组数据和代谢组数据,建立了一系列预后预测模型。
在测试队列中,多模态模型TMPIC结合了多种数据模式,实现了良好的预后预测效果,能够有效区分高风险和低风险的复发患者。该模型包含五个基因、五个代谢物、五个数字病理学特征、免疫组化亚型和临床肿瘤分期。综上所述,该研究建立了一套全面的工作流程,并证实了多模态整合在乳腺癌预后预测中的潜力。
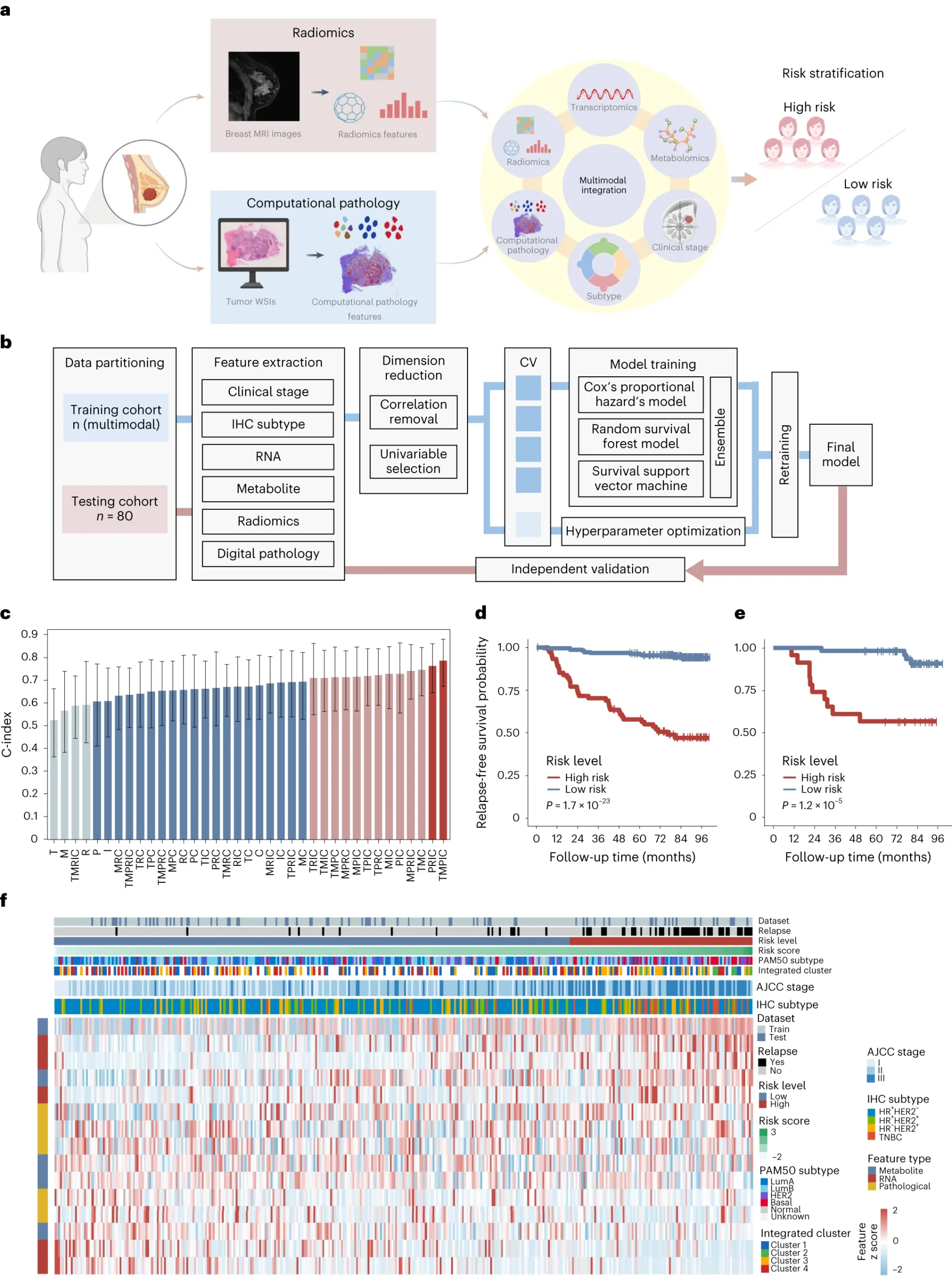
图4 利用机器学习进行多模态数据整合,对乳腺癌进行风险分层
总 结
总的来说,该研究提供了中国人乳腺癌的多组学特征,为未来乳腺癌生物复杂性研究提供了有价值的参考,可能有助于精准治疗的进一步发展。
文章链接:
https://www.nature.com/articles/s43018-024-00725-0