自2010年人类蛋白质组项目(HPP)启动以来, 在过去的22年里,蛋白质组学经历了翻天覆地的变化。从大费周章才能清点一个样品中的近千个蛋白质,到使用高分辨质谱和AI可以轻松对数千个样品的逾万个蛋白质进行定量分析,科学家们已经不再满足于对蛋白质的鉴定和定量。
AI赋能的蛋白质组学能不能带来颠覆性的改变?且看2023年年度报告将会如何书写。
——欧米锐评人 青石
目前,HUPO人类蛋白质组计划在Journal of Proteome Research上在线发表了其在2022年的研究报告,题目为 “The 2022 Report on the Human Proteome from the HUPO Human Proteome Project”。
2022年人类蛋白质组计划(Human Proteome Project,HPP)研究显示,在人类基因组编码的19750种预测蛋白质中,已有效检测到18407种neXtProt PE1(neXtProt Protein existence 1)蛋白质的表达。
自2021年以来,HPP重新分析了来自世界各地的数据又进一步确定了50种能够有效检测的蛋白质。与之相反,neXtProt PE2、PE3 和PE4缺失蛋白质的数量从1421种减少到1343种,减少了78种。这一结果表明了全染色体水平人类蛋白质组的研究进展以及相关重要的蛋白质重新分类情况。
与此同时,蛋白质组学在生物学和临床研究中也有重大发现,并积极与其他多组学平台整合。文中阐述了以染色体为中心的 HPP、生物学和疾病驱动的 HPP 以及 HPP 资源支柱的科研进展,比较了质谱、 Olink 和 Somalogic 平台的特征,关注了核糖体分析中小型开放阅读框出现的翻译产物,讨论了首次启动的HPP Grand Challenge Project— “每个蛋白质都有功能注释” 项目。
♦ 人类蛋白质组的研究进展
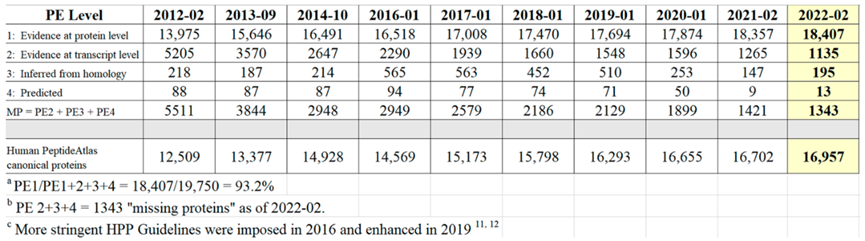
表1 2012-02年至2022-02年neXtProt Protein Existence已验证的蛋白质数量
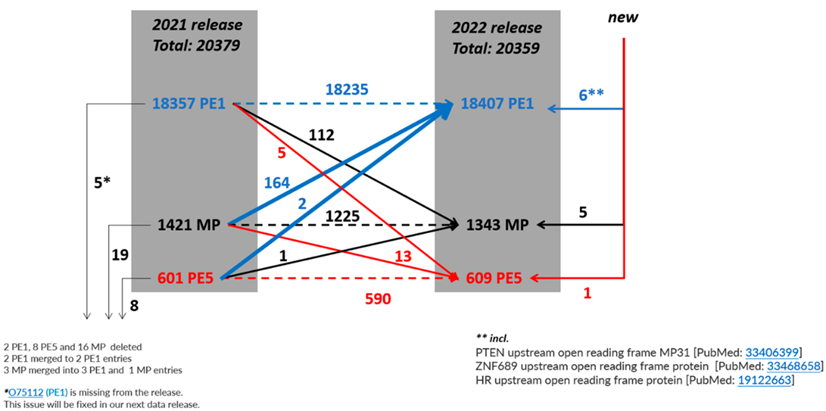
图1 2021-2022年预测蛋白PE1、PE2、PE3、PE4、和PE5数量变化示意图
基于MS鉴定到的PE1数据,neXtProt 结合了来自PeptideAtlas和MassIVE(Mass Spectrometry Interactive Virtual Environment, MassIVE)的肽鉴定图谱,且满足蛋白质验证指南(基于HPP质谱数据解析指南v3.0至少两个特异性肽段的映射,9 个或更多氨基酸的非嵌套肽,覆盖至少18个氨基酸)。在1343个PE2, 3, 4缺失的蛋白质中,有310个MS数据不满足指南。
由图2可知,18407种PE1蛋白质中有17539 种基于质谱数据结果鉴定。其中17174个蛋白质数据来自对PeptideAtlas原始数据的再次整理,17033个来自MassIVE,两者交集共16668种蛋白质在两个库中均符合指南要求。肽类的数据必须至少满足一个数据库的验证指南要求;两个数据库中的部分数据存在不能互相验证的情况。
在868个非MS-based PE1蛋白质中,47个主要基于Edman降解N端蛋白测序,18个基于蛋白质数据库中天然蛋白(非重组蛋白)的3D结构,453个基于蛋白-蛋白相互作用,31个基于抗体研究,87个基于翻译后修饰和蛋白水解处理,69个基于基因突变,163个来自生化研究。许多蛋白质数据来自多种类型研究,如图3所示,其中有些MS数据不足以满足HPP MS数据解析指南v3.0。
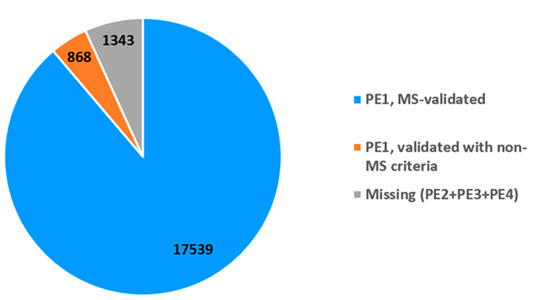
图2 2022年人类蛋白质组中蛋白质的状态
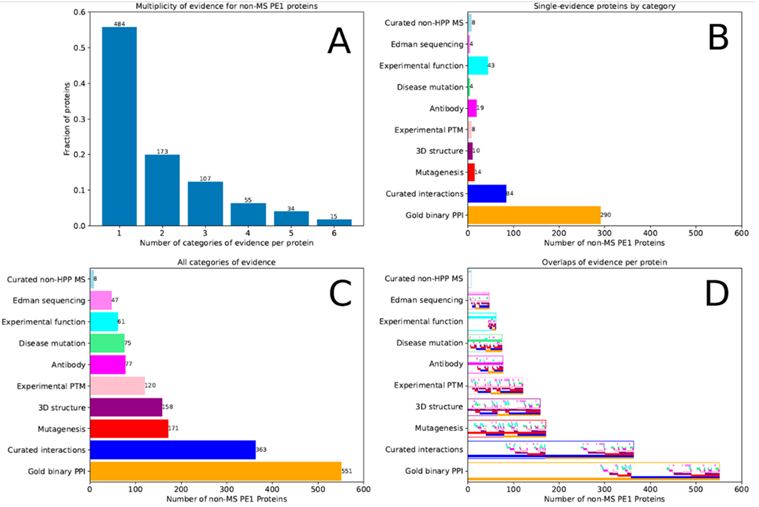
图3 868种不符合PeptideAtlas和MassIVE HPP指南的PE1蛋白
2020-2021年期间,基于之前的蛋白质-蛋白质相互作用提升到PE1的112个条目在2021年至2022年期间有部分被降级,使PE1的净增加减少到50个条目。2016年以来的补充描述如图4所示。
从表1可知,随着有效地识别PE2, 3, 4缺失蛋白方面继续取得显著进展,PE1数量持续增加,同时PE2, 3, 4的数量减少(现在只占PE1,2,3,4总蛋白质的6.8%)。图5显示了在2022年增加的255个蛋白中PeptideAtlas贡献度排名前8的新数据集的160个蛋白质(表1),详见附表S2.
按照目前的鉴定速度来看,至2025年将会得到19250个PE1蛋白质。然而,仍然存在一些难以检测鉴定的蛋白质:其中包括对多达 800 个预测的PE2, 3, 4 蛋白质(基于HPA、GTEx、FAN-TOM5);部分溶解度不好的膜蛋白;仅在未研究的组织或细胞类型中表达的蛋白质;缺少胰蛋白酶消化位点(两个相距 9-40 个氨基酸)的蛋白质(尽管这些蛋白质可以用替代蛋白酶、缺失的切割或末端肽检测)。
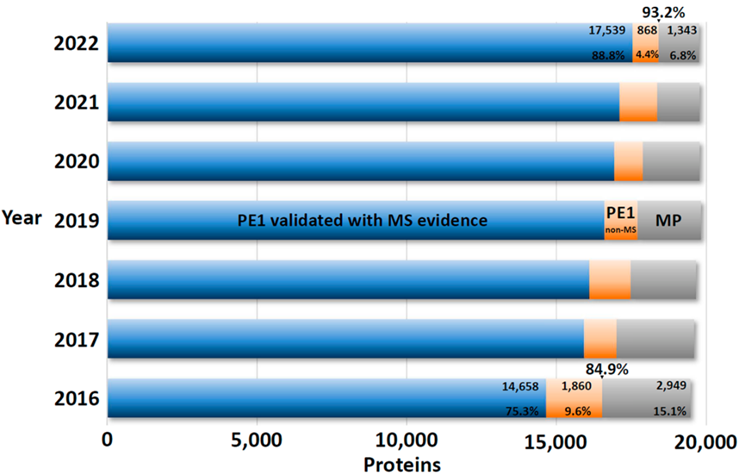
图4 PE2, 3, 4蛋白质(灰色)和PE1蛋白质(蓝色+橙色)鉴定进展

图5 在Protein Atlas 2022-01和neXtProt 2022-02中生成10个或以上新PE1蛋白质的主要论文
♦ 以染色体为中心的HPP研究进展(THE CHROMOSOME-CENTRIC-HPP, C-HPP)
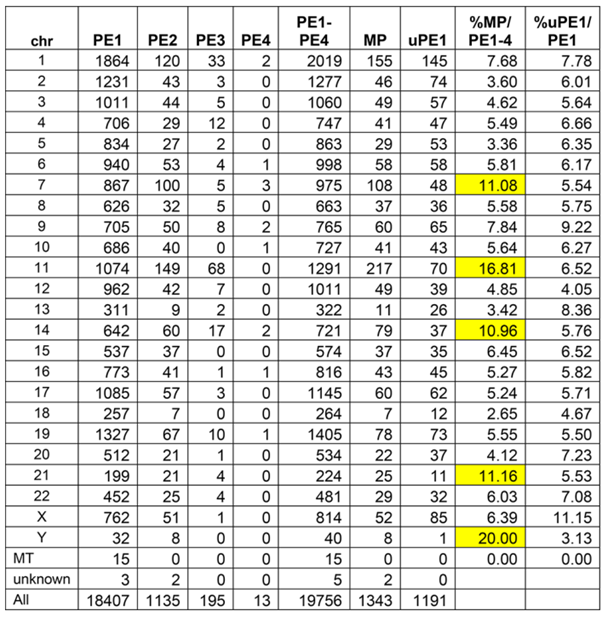
表2 neXtProt 2022-02染色体上蛋白质的预测情况
♦ 生物和疾病驱动的B/D-HPP研究进展(The Biology and Disease-driven Human Proteome Project,B/D-HPP)
◊心血管疾病
◊癌症
◊糖蛋白质组学
◊人类免疫肽组研究
◊食品和营养
◊风湿性和自身免疫性疾病
◊传染性疾病
♦ 四个资源支柱的发展情况
4.1 知识库资源支柱研究进展
从 Ribo-seq Open Reading Frame (ORF) 序列翻译潜在多肽
4.2 抗体支柱/人类蛋白质图谱研究进展
4.3 病理资源支柱研究进展
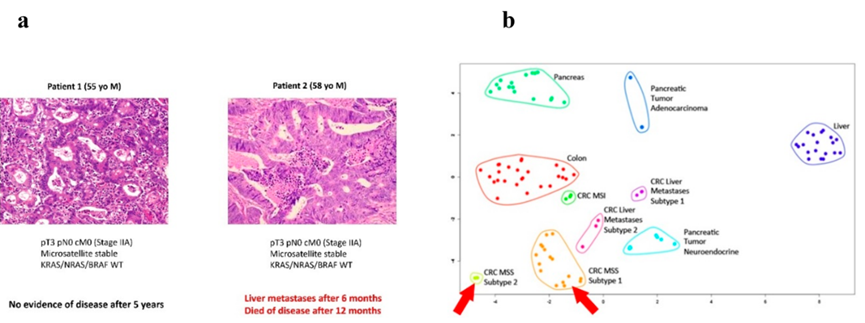
图6 基于蛋白质组学的结肠癌个性化结果风险预测
最后,HPP病理学的使命是通过会议和辅导,加强对全世界医生和医疗机构的教育和宣传,提高对蛋白质组学的认识并促进国际病理学和实验室医学学会与HUPO和HPP建立密切的合作联系,将这些举措推广到世界各地。
血液中蛋白质鉴定的亲和力测定法的不断发展,加速了这些技术在高通量血浆和血清蛋白质组学中的应用。最近对这些基于非质谱技术的血液蛋白质组学技术的评论描述了每种技术的优点和缺点,并强调需要对每种技术进行定量评估和比较以验证每种方法。
靶向检测发展中最突出的是近似延伸检测(Olink公司的PEA),其构造为成对的抗体,以定量聚合酶链反应(qPCR)或测序作为读数,或大量的改性慢速DNA适配体库(SomaLogic的SomaScan)。Olink提供了2940种(3K系统)独特的检测方法,SomaLogic提供了6377种(7K系统)针对人类血浆蛋白的检测方法。
目前,这些平台间目标蛋白质重叠度过高结合所有三个平台,有超过8000种蛋白质可以作为潜在检测血浆蛋白质的目标。
过去几年,有研究已经比较了不同的靶向检测方法及其检出的关联性。但是这些大规模的靶向测定只报告相对的定量值,且靶向蛋白质结合分子的方法不同,每个被测定的蛋白质的结合表位不同,以及检测方式不同,因此在测定之间的关联反应有相当大的困难。相关性差异很大,对不同平台的研究结果难以关联的解释。
这些差异使需要准确鉴定和定量的临床化学应用变得非常复杂。开发一项新的技术,需要很多实验证据验证其特异性和准确性。Somalogic公司评估了他们基于诱导剂的亲和试剂的特异性,虽然在这项实验中每种试剂的定量准确性还有待调查,但这组数据为确定这些新技术之一的核心试剂的特征提供了第一个证据。
利用标准化的96孔板靶向蛋白质组学测量的高通量分析,可以快速量化实验中和不同实验中检测到的分析物的差异。在SCALLOP内,拥有Olink数据的研究人员希望比较不同队列的结果,从而增加可搜索结果的总库。
目前,SCALLOP有超过70000个样本结果可供成员使用。另一个大型联盟是人类制药蛋白质组学项目(HPPP),最近完成了对英国生物库参与者的54306份血浆蛋白质组图谱的初步描述。使用Olink 1.5K panel(1463个独特的蛋白质)作为第一阶段的分析,并包括蛋白质定量性状位点(pQTL)图谱,确定了10248个初级遗传关联,其中85%是新发现的。数据确定了92%的顺式位点和29%的反式位点的独立二级关联,扩大了用于下游分析的遗传工具目录。
这项研究提供了血浆蛋白质组遗传结构的最新特征,利用群体规模的蛋白质组学为跨多个生物领域的反式pQTLs提供新的广泛结果。
这项大型研究用一种单一的技术确定了许多可操作的药理结果,如确定对配体-受体相互作用的遗传影响、通路扰动和新的药物靶点,PCSK9水平对脂质浓度和心脑血管疾病的遗传代理效应。
这些数据将Olink分析扩展到Olink 3K panel,并在较小的UKB受试者群中进行各种基于质谱的试验规模分析。将公共血浆蛋白质组知识库作为一种开放的蛋白质组学资源,将有助于阐明遗传发现背后的生物机制,并加速新型生物标志物和治疗方法的开发。为了使平台之间的定量反应相关化,在Price、Ruffieux这两个团队的系统研究中将质谱法与Olink检测的定量读数进行了比较。
基于质谱的方法并没有摆脱定量的困境,质谱中绝大多数的蛋白质检测和定量都来自于自下而上的方法,其中蛋白质被酶切成短肽,在质谱仪中被鉴定和定量,然后最后汇总成一个蛋白质水平的定量值。鉴于每个基因产物估计有100种不同的蛋白形式,肽信号的平均化不考虑翻译后修饰的贡献,可能会妨碍发现重要的生物差异。
在我们了解每个被鉴定的蛋白质的背景之前,任何技术所提供的每个蛋白质的定量值都需要用正交的方法来验证,以说明生物的多样性。随着技术的不断发展,需要利用每种技术的优势,并以跨平台的能力作为补充,以得出解释生物差异的结论。
♦ HPP Grand Challenge Project:“每个蛋白质都有功能注释”
原文链接:https://doi.org/10.1021/acs.jproteome.2c00498
编译:王佳童
自2010年人类蛋白质组项目(HPP)启动以来, 在过去的22年里,蛋白质组学经历了翻天覆地的变化。从大费周章才能清点一个样品中的近千个蛋白质,到使用高分辨质谱和AI可以轻松对数千个样品的逾万个蛋白质进行定量分析,科学家们已经不再满足于对蛋白质的鉴定和定量。
AI赋能的蛋白质组学能不能带来颠覆性的改变?且看2023年年度报告将会如何书写。
——欧米锐评人 青石
目前,HUPO人类蛋白质组计划在Journal of Proteome Research上在线发表了其在2022年的研究报告,题目为 “The 2022 Report on the Human Proteome from the HUPO Human Proteome Project”。
2022年人类蛋白质组计划(Human Proteome Project,HPP)研究显示,在人类基因组编码的19750种预测蛋白质中,已有效检测到18407种neXtProt PE1(neXtProt Protein existence 1)蛋白质的表达。
自2021年以来,HPP重新分析了来自世界各地的数据又进一步确定了50种能够有效检测的蛋白质。与之相反,neXtProt PE2、PE3 和PE4缺失蛋白质的数量从1421种减少到1343种,减少了78种。这一结果表明了全染色体水平人类蛋白质组的研究进展以及相关重要的蛋白质重新分类情况。
与此同时,蛋白质组学在生物学和临床研究中也有重大发现,并积极与其他多组学平台整合。文中阐述了以染色体为中心的 HPP、生物学和疾病驱动的 HPP 以及 HPP 资源支柱的科研进展,比较了质谱、 Olink 和 Somalogic 平台的特征,关注了核糖体分析中小型开放阅读框出现的翻译产物,讨论了首次启动的HPP Grand Challenge Project— “每个蛋白质都有功能注释” 项目。
♦ 人类蛋白质组的研究进展
表1 2012-02年至2022-02年neXtProt Protein Existence已验证的蛋白质数量
图1 2021-2022年预测蛋白PE1、PE2、PE3、PE4、和PE5数量变化示意图
基于MS鉴定到的PE1数据,neXtProt 结合了来自PeptideAtlas和MassIVE(Mass Spectrometry Interactive Virtual Environment, MassIVE)的肽鉴定图谱,且满足蛋白质验证指南(基于HPP质谱数据解析指南v3.0至少两个特异性肽段的映射,9 个或更多氨基酸的非嵌套肽,覆盖至少18个氨基酸)。在1343个PE2, 3, 4缺失的蛋白质中,有310个MS数据不满足指南。
由图2可知,18407种PE1蛋白质中有17539 种基于质谱数据结果鉴定。其中17174个蛋白质数据来自对PeptideAtlas原始数据的再次整理,17033个来自MassIVE,两者交集共16668种蛋白质在两个库中均符合指南要求。肽类的数据必须至少满足一个数据库的验证指南要求;两个数据库中的部分数据存在不能互相验证的情况。
在868个非MS-based PE1蛋白质中,47个主要基于Edman降解N端蛋白测序,18个基于蛋白质数据库中天然蛋白(非重组蛋白)的3D结构,453个基于蛋白-蛋白相互作用,31个基于抗体研究,87个基于翻译后修饰和蛋白水解处理,69个基于基因突变,163个来自生化研究。许多蛋白质数据来自多种类型研究,如图3所示,其中有些MS数据不足以满足HPP MS数据解析指南v3.0。
图2 2022年人类蛋白质组中蛋白质的状态
图3 868种不符合PeptideAtlas和MassIVE HPP指南的PE1蛋白
2020-2021年期间,基于之前的蛋白质-蛋白质相互作用提升到PE1的112个条目在2021年至2022年期间有部分被降级,使PE1的净增加减少到50个条目。2016年以来的补充描述如图4所示。
从表1可知,随着有效地识别PE2, 3, 4缺失蛋白方面继续取得显著进展,PE1数量持续增加,同时PE2, 3, 4的数量减少(现在只占PE1,2,3,4总蛋白质的6.8%)。图5显示了在2022年增加的255个蛋白中PeptideAtlas贡献度排名前8的新数据集的160个蛋白质(表1),详见附表S2.
按照目前的鉴定速度来看,至2025年将会得到19250个PE1蛋白质。然而,仍然存在一些难以检测鉴定的蛋白质:其中包括对多达 800 个预测的PE2, 3, 4 蛋白质(基于HPA、GTEx、FAN-TOM5);部分溶解度不好的膜蛋白;仅在未研究的组织或细胞类型中表达的蛋白质;缺少胰蛋白酶消化位点(两个相距 9-40 个氨基酸)的蛋白质(尽管这些蛋白质可以用替代蛋白酶、缺失的切割或末端肽检测)。
图4 PE2, 3, 4蛋白质(灰色)和PE1蛋白质(蓝色+橙色)鉴定进展
图5 在Protein Atlas 2022-01和neXtProt 2022-02中生成10个或以上新PE1蛋白质的主要论文
♦ 以染色体为中心的HPP研究进展(THE CHROMOSOME-CENTRIC-HPP, C-HPP)
表2 neXtProt 2022-02染色体上蛋白质的预测情况
♦ 生物和疾病驱动的B/D-HPP研究进展(The Biology and Disease-driven Human Proteome Project,B/D-HPP)
◊心血管疾病
◊癌症
◊糖蛋白质组学
◊人类免疫肽组研究
◊食品和营养
◊风湿性和自身免疫性疾病
◊传染性疾病
♦ 四个资源支柱的发展情况
4.1 知识库资源支柱研究进展
从 Ribo-seq Open Reading Frame (ORF) 序列翻译潜在多肽
4.2 抗体支柱/人类蛋白质图谱研究进展
4.3 病理资源支柱研究进展
图6 基于蛋白质组学的结肠癌个性化结果风险预测
最后,HPP病理学的使命是通过会议和辅导,加强对全世界医生和医疗机构的教育和宣传,提高对蛋白质组学的认识并促进国际病理学和实验室医学学会与HUPO和HPP建立密切的合作联系,将这些举措推广到世界各地。
血液中蛋白质鉴定的亲和力测定法的不断发展,加速了这些技术在高通量血浆和血清蛋白质组学中的应用。最近对这些基于非质谱技术的血液蛋白质组学技术的评论描述了每种技术的优点和缺点,并强调需要对每种技术进行定量评估和比较以验证每种方法。
靶向检测发展中最突出的是近似延伸检测(Olink公司的PEA),其构造为成对的抗体,以定量聚合酶链反应(qPCR)或测序作为读数,或大量的改性慢速DNA适配体库(SomaLogic的SomaScan)。Olink提供了2940种(3K系统)独特的检测方法,SomaLogic提供了6377种(7K系统)针对人类血浆蛋白的检测方法。
目前,这些平台间目标蛋白质重叠度过高结合所有三个平台,有超过8000种蛋白质可以作为潜在检测血浆蛋白质的目标。
过去几年,有研究已经比较了不同的靶向检测方法及其检出的关联性。但是这些大规模的靶向测定只报告相对的定量值,且靶向蛋白质结合分子的方法不同,每个被测定的蛋白质的结合表位不同,以及检测方式不同,因此在测定之间的关联反应有相当大的困难。相关性差异很大,对不同平台的研究结果难以关联的解释。
这些差异使需要准确鉴定和定量的临床化学应用变得非常复杂。开发一项新的技术,需要很多实验证据验证其特异性和准确性。Somalogic公司评估了他们基于诱导剂的亲和试剂的特异性,虽然在这项实验中每种试剂的定量准确性还有待调查,但这组数据为确定这些新技术之一的核心试剂的特征提供了第一个证据。
利用标准化的96孔板靶向蛋白质组学测量的高通量分析,可以快速量化实验中和不同实验中检测到的分析物的差异。在SCALLOP内,拥有Olink数据的研究人员希望比较不同队列的结果,从而增加可搜索结果的总库。
目前,SCALLOP有超过70000个样本结果可供成员使用。另一个大型联盟是人类制药蛋白质组学项目(HPPP),最近完成了对英国生物库参与者的54306份血浆蛋白质组图谱的初步描述。使用Olink 1.5K panel(1463个独特的蛋白质)作为第一阶段的分析,并包括蛋白质定量性状位点(pQTL)图谱,确定了10248个初级遗传关联,其中85%是新发现的。数据确定了92%的顺式位点和29%的反式位点的独立二级关联,扩大了用于下游分析的遗传工具目录。
这项研究提供了血浆蛋白质组遗传结构的最新特征,利用群体规模的蛋白质组学为跨多个生物领域的反式pQTLs提供新的广泛结果。
这项大型研究用一种单一的技术确定了许多可操作的药理结果,如确定对配体-受体相互作用的遗传影响、通路扰动和新的药物靶点,PCSK9水平对脂质浓度和心脑血管疾病的遗传代理效应。
这些数据将Olink分析扩展到Olink 3K panel,并在较小的UKB受试者群中进行各种基于质谱的试验规模分析。将公共血浆蛋白质组知识库作为一种开放的蛋白质组学资源,将有助于阐明遗传发现背后的生物机制,并加速新型生物标志物和治疗方法的开发。为了使平台之间的定量反应相关化,在Price、Ruffieux这两个团队的系统研究中将质谱法与Olink检测的定量读数进行了比较。
基于质谱的方法并没有摆脱定量的困境,质谱中绝大多数的蛋白质检测和定量都来自于自下而上的方法,其中蛋白质被酶切成短肽,在质谱仪中被鉴定和定量,然后最后汇总成一个蛋白质水平的定量值。鉴于每个基因产物估计有100种不同的蛋白形式,肽信号的平均化不考虑翻译后修饰的贡献,可能会妨碍发现重要的生物差异。
在我们了解每个被鉴定的蛋白质的背景之前,任何技术所提供的每个蛋白质的定量值都需要用正交的方法来验证,以说明生物的多样性。随着技术的不断发展,需要利用每种技术的优势,并以跨平台的能力作为补充,以得出解释生物差异的结论。
♦ HPP Grand Challenge Project:“每个蛋白质都有功能注释”
原文链接:https://doi.org/10.1021/acs.jproteome.2c00498
编译:王佳童