







新药研发是一项漫长、昂贵和低效的系统工程,制药行业的反摩尔定律使得药物发现的成本越来越高、成功率不断下降。
2020年,“人工智能辅助药物设计”入选《麻省理工科技评论》2020年度“全球十大突破性技术”。现代人工智能的发展,给药物研发带来了新的曙光。
作为新药研发的入口,药物发现是目前AI应用得最多也最成熟的环节。据统计,AI能将药物发现时间缩短40%。AI的深度学习领域可以辅助药物发现的多种应用,例如生成小分子结构、辅助类药化合物设计、预测小分子与靶标的作用、筛选化合物与发现先导物等。
本月初,意大利学者Carmen Cerchia等人发表的新文章讨论了AI辅助药物发现的最新技术和示范应用,例如基于生成模型的药物分子设计、研究蛋白质-配体结合过程等。这些研究对AI辅助药物设计有着深远影响,也带来了新的挑战。本文梳理了这篇文章的主要内容,概述了应用于药物发现的最相关的机器学习方法。
—— 欧米锐评人 林木
本文原载于《智药邦》,《西湖欧米》获权转载。
2023年2月2日,意大利那不勒斯大学的 Carmen Cerchia 等人在 Drug Discovery Today上发表文章New avenues in artificial-intelligence-assisted drug discovery。

作者讨论了人工智能(AI)辅助药物发现的当前最新技术、最近的应用,包括用于化学结构生成的生成模型、改善结合亲和力和姿势预测的评分函数,以及协助参数化、特征化和泛化任务的分子动力学等内容。
♦ 背景
深度学习的出现在解决药物发现中的各种问题(如从头分子设计)方面显示出巨大的前景。从头设计技术已被大量利用,为药物发现活动提供了有希望的起点。从头开始的药物设计基本上是指自动构建具有理想分子结构的新化学结构的过程。近年来,结合生成式模型的分子设计越来越受欢迎。
♦ 生成式模型用于药物设计
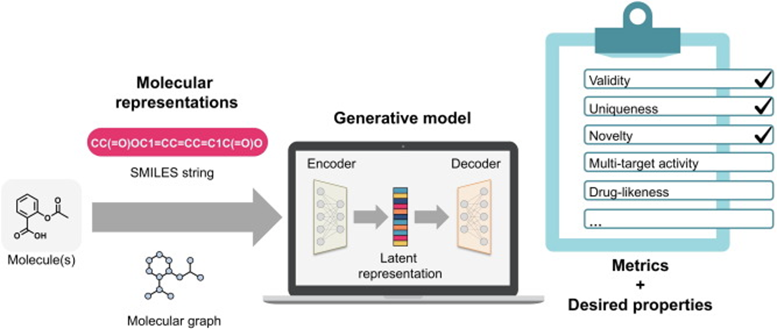
图1 生成式模型用于药物设计输入分子表示在控制模型学习阶段(即,模型如何获得分子信息)中具有关键作用。有三种主要表示:(i)1D(例如字符串);(ii)2D(例如分子图);以及(iii)3D(例如坐标)。
♦ 基于药物-蛋白质复合物的分子生成
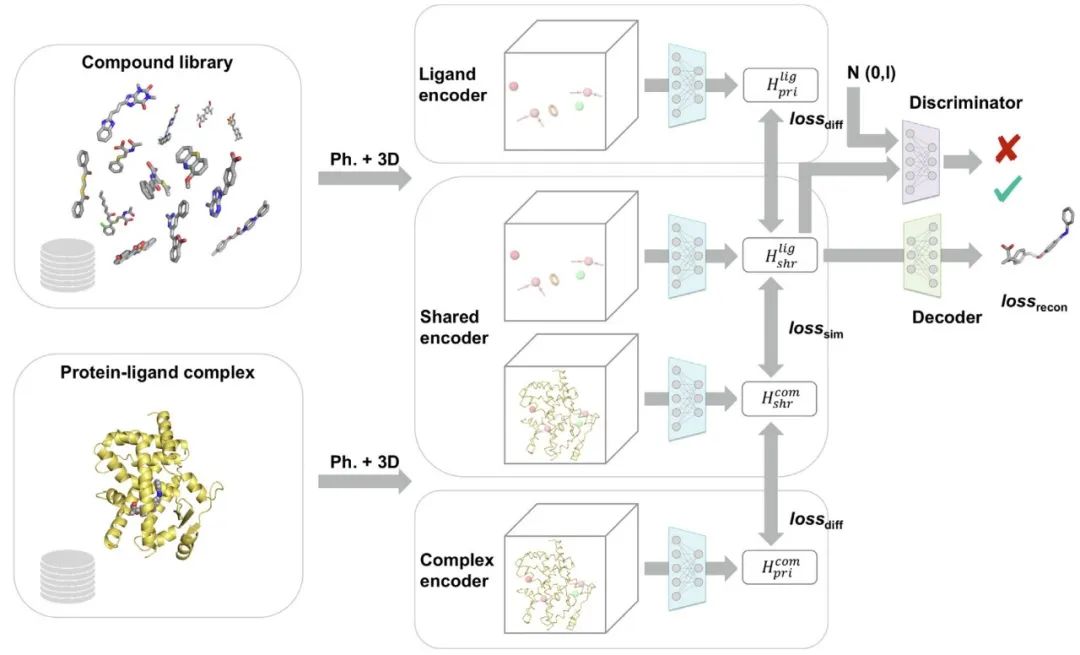
图2 RELATION模型
♦ 总结
参考资料
[1] Cerchia et al. New avenues in artificial-intelligence-assisted drug discovery. Drug Discov Today. 2023
[2] Jin et al. Hierarchical Generation of Molecular Graphs using Structural Motifs. ICML. 2020
[3] Axelrod et al. GEOM: Energy-annotated molecular conformations for property prediction and molecular generation. Sci Data. 2022
[4] Gomez-Bombarelli et al. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent Sci. 2018
[5] Zhavoronkov et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat Biotechnol. 2019
[6] Li et al. Multi-objective de novo drug design with conditional graph generative model. J Cheminform. 2018
[7] Fu et al. MIMOSA: Multi-constraint Molecule Sampling for Molecule Optimization. AAAI. 2020
[8] Bung et al. An In Silico Explainable Multiparameter Optimization Approach for De Novo Drug Design against Proteins from the Central Nervous System. J Chem Inf Model. 2022
[9] Ragoza, et al. Generating 3D molecules conditional on receptor binding sites with deep generative models. Chem. Sci. 2022
[10] Wang, et al. RELATION: A Deep Generative Model for Structure-Based De Novo Drug Design. J. Med. Chem. 2022
[11] Walters et al. Assessing the impact of generative AI on medicinal chemistry. Nat Biotechnol. 2020
新药研发是一项漫长、昂贵和低效的系统工程,制药行业的反摩尔定律使得药物发现的成本越来越高、成功率不断下降。
2020年,“人工智能辅助药物设计”入选《麻省理工科技评论》2020年度“全球十大突破性技术”。现代人工智能的发展,给药物研发带来了新的曙光。
作为新药研发的入口,药物发现是目前AI应用得最多也最成熟的环节。据统计,AI能将药物发现时间缩短40%。AI的深度学习领域可以辅助药物发现的多种应用,例如生成小分子结构、辅助类药化合物设计、预测小分子与靶标的作用、筛选化合物与发现先导物等。
本月初,意大利学者Carmen Cerchia等人发表的新文章讨论了AI辅助药物发现的最新技术和示范应用,例如基于生成模型的药物分子设计、研究蛋白质-配体结合过程等。这些研究对AI辅助药物设计有着深远影响,也带来了新的挑战。本文梳理了这篇文章的主要内容,概述了应用于药物发现的最相关的机器学习方法。
—— 欧米锐评人 林木
本文原载于《智药邦》,《西湖欧米》获权转载。
2023年2月2日,意大利那不勒斯大学的 Carmen Cerchia 等人在 Drug Discovery Today上发表文章New avenues in artificial-intelligence-assisted drug discovery。
作者讨论了人工智能(AI)辅助药物发现的当前最新技术、最近的应用,包括用于化学结构生成的生成模型、改善结合亲和力和姿势预测的评分函数,以及协助参数化、特征化和泛化任务的分子动力学等内容。
♦ 背景
深度学习的出现在解决药物发现中的各种问题(如从头分子设计)方面显示出巨大的前景。从头设计技术已被大量利用,为药物发现活动提供了有希望的起点。从头开始的药物设计基本上是指自动构建具有理想分子结构的新化学结构的过程。近年来,结合生成式模型的分子设计越来越受欢迎。
♦ 生成式模型用于药物设计
图1 生成式模型用于药物设计输入分子表示在控制模型学习阶段(即,模型如何获得分子信息)中具有关键作用。有三种主要表示:(i)1D(例如字符串);(ii)2D(例如分子图);以及(iii)3D(例如坐标)。
♦ 基于药物-蛋白质复合物的分子生成
图2 RELATION模型
♦ 总结
参考资料
[1] Cerchia et al. New avenues in artificial-intelligence-assisted drug discovery. Drug Discov Today. 2023
[2] Jin et al. Hierarchical Generation of Molecular Graphs using Structural Motifs. ICML. 2020
[3] Axelrod et al. GEOM: Energy-annotated molecular conformations for property prediction and molecular generation. Sci Data. 2022
[4] Gomez-Bombarelli et al. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent Sci. 2018
[5] Zhavoronkov et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat Biotechnol. 2019
[6] Li et al. Multi-objective de novo drug design with conditional graph generative model. J Cheminform. 2018
[7] Fu et al. MIMOSA: Multi-constraint Molecule Sampling for Molecule Optimization. AAAI. 2020
[8] Bung et al. An In Silico Explainable Multiparameter Optimization Approach for De Novo Drug Design against Proteins from the Central Nervous System. J Chem Inf Model. 2022
[9] Ragoza, et al. Generating 3D molecules conditional on receptor binding sites with deep generative models. Chem. Sci. 2022
[10] Wang, et al. RELATION: A Deep Generative Model for Structure-Based De Novo Drug Design. J. Med. Chem. 2022
[11] Walters et al. Assessing the impact of generative AI on medicinal chemistry. Nat Biotechnol. 2020


