♦ AI揭示多组学数据与药物关联

图源:论文截图
图源:文章图片
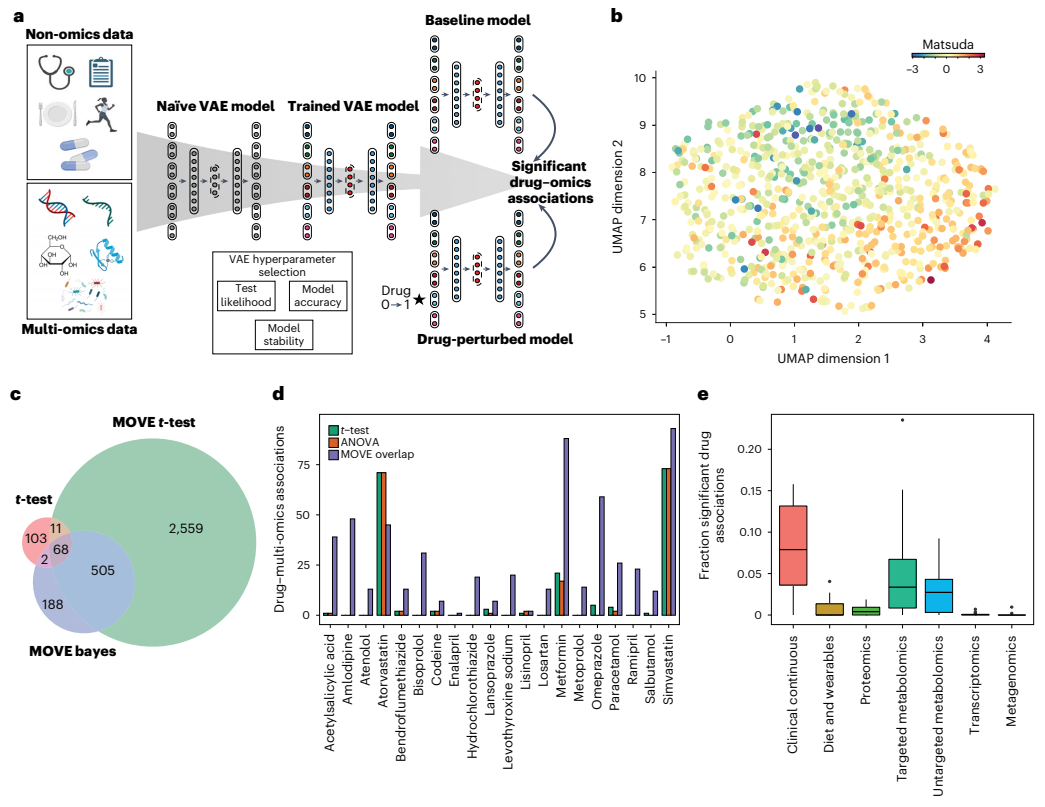
利用多组学分析技术对临床队列进行分析有可能揭示出患者水平的疾病特征以及分析个性化的治疗反应。但如何结合各个组学(即多模态)的数据是一个具有挑战的任务。为了更好的整合多组学的数据,作者基于传统深度学习模型VAE搭建了多组学变分编码器(MOVE),并将模型应用于789名2型糖尿病患者队列,模型输入为个体水平的非组学、多组学数据以及药物状态的有无,以此来确定药物和组学的关联性。为了增强模型的可解释性,对神经网络的权重进行分析以及SHAP方法研究特征各个特征是如何影响个体在空间中的定位。进一步,作者模拟了一次扰动的特征输入方法以及新的分析方法(MOVE t-test、MOVE Bayes)来评估模型是否学习到了药物和多组学之间的关系,结果表明两种分析方法表现良好,其FDR为0.05。随后,团队应用MOVE框架识别DIRECT数据集中的药物关联,发现T2D生物标志物的变化与二甲双胍有关、二甲双胍和奥美拉唑与肠道菌群存在一定关系。同时还发现了一些药物引起组学变化之间的相似性。该研究不仅可以发现新的生物标志物,还可以从多组学数据上从患者层面进行药物选择。该研究为多组学的融合分析提供了一种可行的思路,同时,作者也提出了未来继续融合其它组学数据的期望。
https://www.nature.com/articles/s41587-022-01520-x
♦ 多模态分析方法

图源:论文截图
图源:文章图片
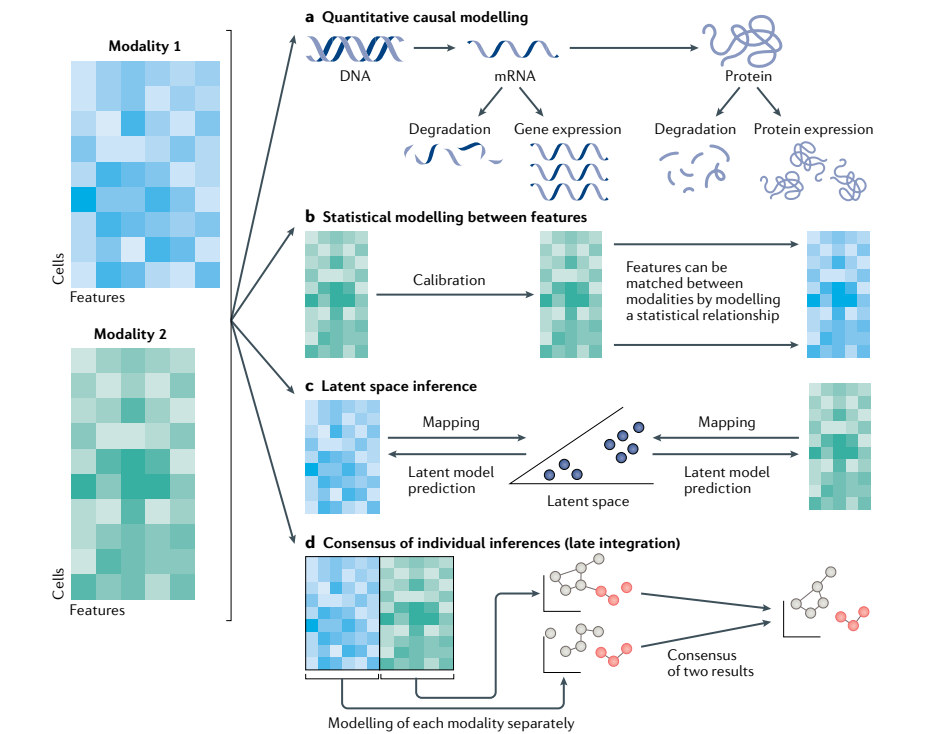
自2015年以来已开发30多项单细胞技术,然而数据整合面临多维度/高噪音/海量数据多方面问题。作者详述了多组学尤其是转录组与蛋白组多模态映射的相关算法及各自的优缺点(如将转录组和蛋白组进行关联映射的CiteFuse)。即便是非同一细胞来源的多组学数据分析依然在临床研究中具有重要意义(如揭示慢性肾病中肌成纤维细胞活化的机制)。作者乐观估计未来海量数据多模态联合分析将发挥更大作用。
https://www.nature.com/articles/s41581-021-00463-x
♦ 一种基于浏览器的开源 ML 工具

图源:论文截图
图源:文章Abstract
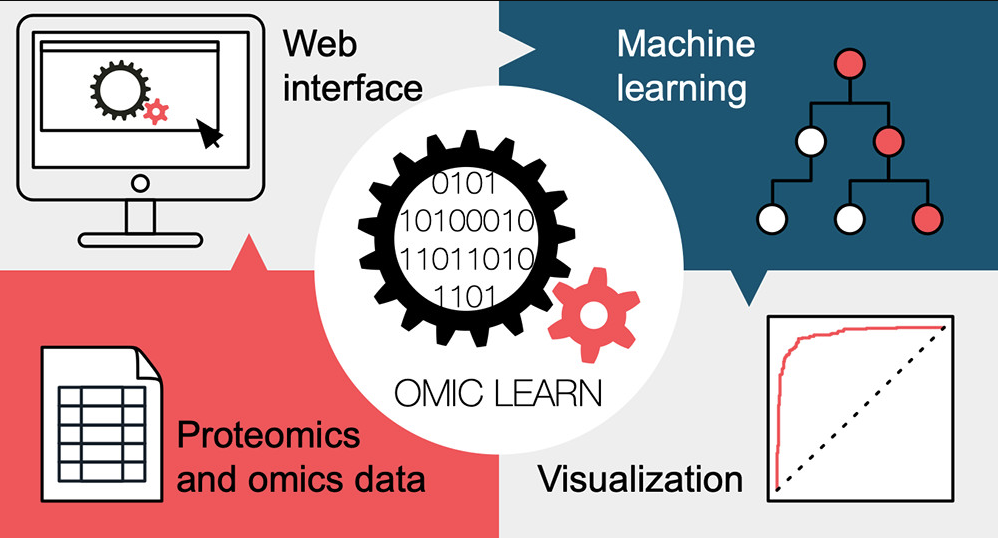
哥本哈根大学Strauss课题组开发了“OmicLearn”(http://OmicLearn.org):一种基于浏览器的开源 ML 工具,可以快速探索各种 ML 算法对实验数据集的适用性。可以在线使用或者安装在本地。他们利用 OmicLearn 来评估基于 ML 的分类将患者与对照组区分开来的程度。OmicLearn 将数据反复拆分为不同的子集,训练分类器并提供性能指标。所得 ROC 曲线的平均曲线下面积 (AUC) 为 0.90 ± 0.08,PR 曲线的平均 AUC 为 0.92 ± 0.06。支持探索性数据分析(PCA、层次聚类)、数据预处理(若干归一化算法)、特征选择(如极度随机树)、分类算法(LinearSVC、AdaBoost等)以及交叉验证算法。通过该工具可以不用编程的实现生物标记物的发现。
♦ Science首席Editor对可以自动写文章问答系统ChatGPT的评论
图源:文章截图
Science 的首席Editor H. Holden Thorp本周评论了关于最近很火的可以自动写文章问答系统的ChatGPT ,强调Sceince系列期刊是不接受使用AI工具如ChatGPT写的文章的:这种基于AI自动生成文章因为产生自机器而不能证明“该作品是原创的”,相当于一种抄袭的。他们目前正在改Editorial Policies, 加一个rules表明science杂志不能接收使用由 ChatGPT(或任何其他人工智能工具)生成的文本,数字、图像或图形文章,反这些政策将将构成科学不端行为, 他们认为这和与篡改图像或抄袭现有作品无异。AI机器的确发挥着重要作用,但它只是人们提出假设、设计实验方法和结讨论果的工具。最终的科研著作必须来自我们头脑中的“计算机”来表达。
笔者认为他们对AI抄袭的定义其实是对AI机器没有真人的贡献。我们可以采取中和的方式,一部分AI一部分自己写,根据我们自己的文章生成一个对话系统写初稿,然后再结合自己的实验结果修改,这种文章,至少还是避免了一些大框架逻辑的错误和语言的错误,还是提高了一部分效率。
https://www.science.org/doi/10.1126/science.adg7879
评论:张芳菲
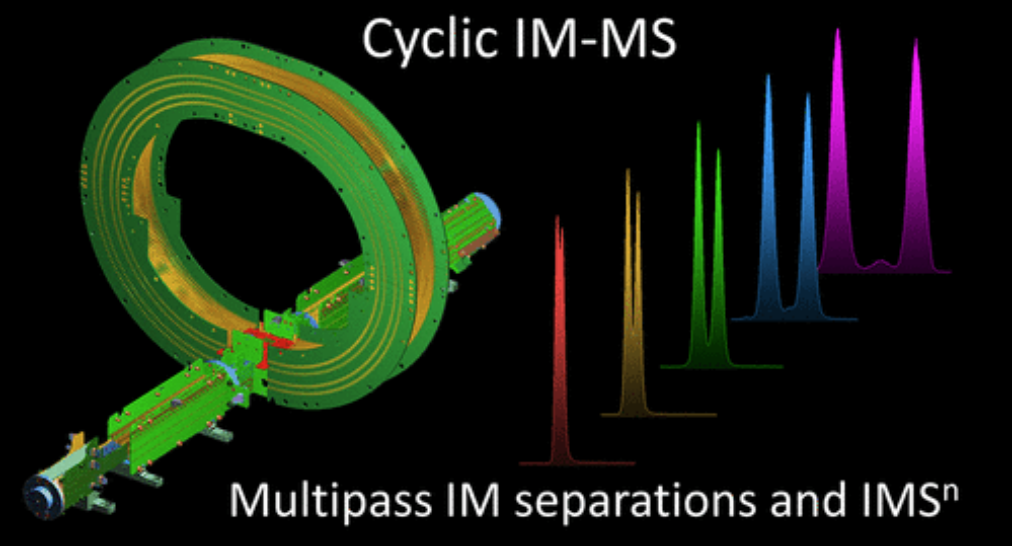
♦ Cyclic Ion Mobility-Mass Spectrometry技术
图源:论文1截图

图源:论文2截图
图源:文章Abstract
第一篇文章介绍了Cyclic Ion Mobility-Mass Spectrometry技术,在传统的IM-Q-TOF仪器上,用98cm长的的圆形行波离子淌度池替代线性离子淌度池,通过循环装置扩展了离子淌度分离的优势。数据显示,通过3次循环分离,5种寡糖(拥有同分异构体的前体离子和碎片离子)可以很好地区分,显示了其在同分异构体的区分上有非常大的潜力。
第二篇文章介绍了基于该技术成功分离了阿尔兹海默症病人的脑组织样本中tau蛋白的异构磷酸肽,这对神经性退行性疾病的病理性蛋白修饰的研究提供了一个可行性的分析流程。
♦ 基于分子互作网络和开源数据库建立疾病特异性的药物蛋白连接谱
图源:论文截图
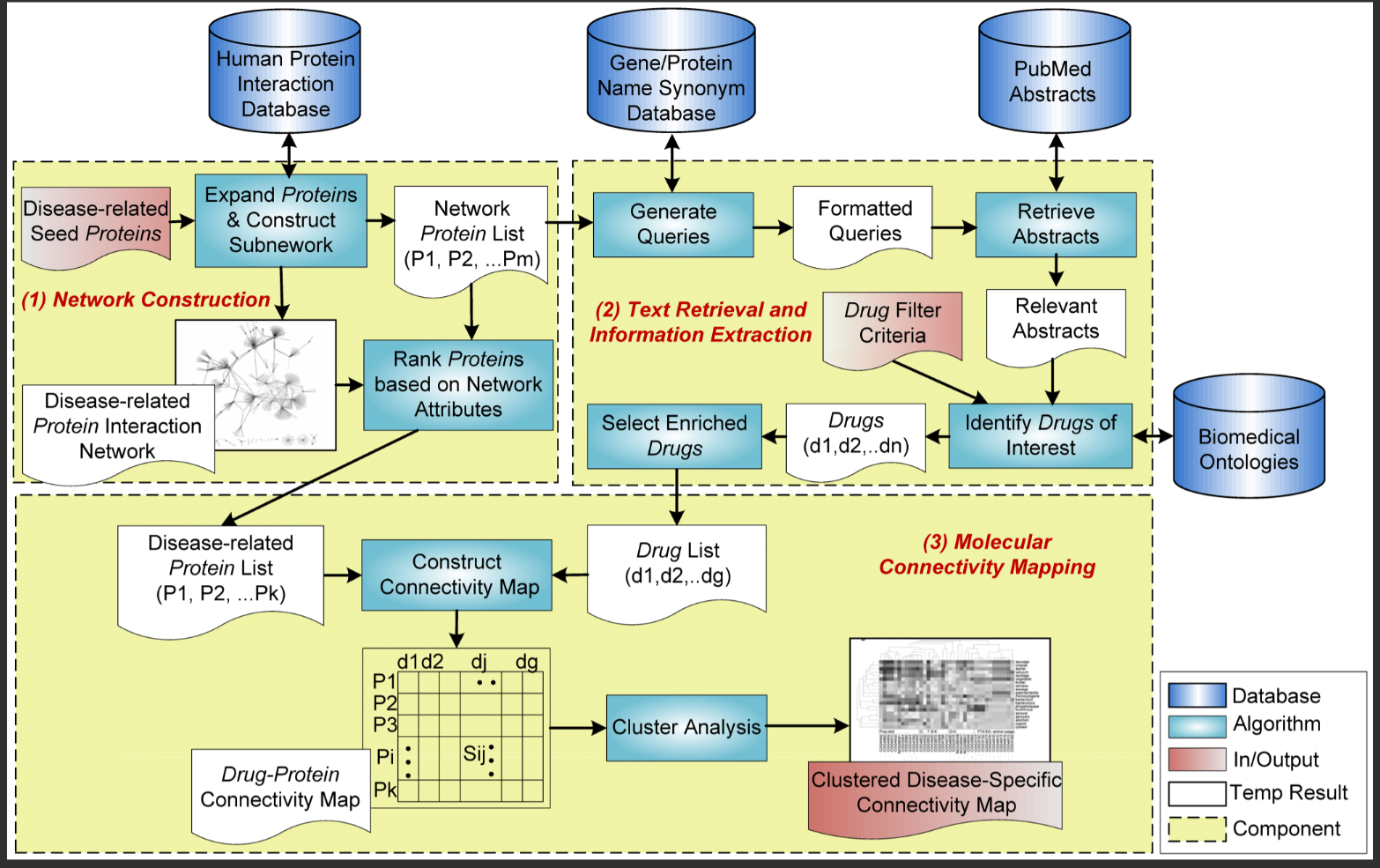
图源:文章Figure
随着基因-药物的分子连接图谱揭示越来越多的基因和药物化合物之间的生物计量关系,如何针对特定疾病提供具有临床应用价值的药物化合物信息,2009年时产生了一次针对疾病-蛋白-药物连接图整合的计算尝试,在没有生成蛋白质-基因-药物化合物连接图的年代,通过整合
1)蛋白相互作用网络,
2)蛋白-基因名检索及同义词扩展,匹配pubmed摘要中提到的药物名称
3)生成疾病相关蛋白与药物化合物矩阵,在此基础上进行过滤和聚类分析。该文章以AD为例子,展示了一个针对AD的计算分子(蛋白-药物)连接图,并筛选出地尔硫卓和奎尼丁肯可能成为作为AD治疗的候选药物。截至目前,该文章共引用两百余次,但很遗憾的是,跟据该方法搭建的支持搜索疾病相关的蛋白-药物关系的网站已经不再维护。结合GUOmics的研究,可以推广到,如果我们拥有实验得到的蛋白-药物连接图,可以整合CORUM,Bioplex等通过实验得到的complex resource,通过in silico得到蛋白质复合物-蛋白-药物化合物之间的连接图。
♦ 一种卷积神经网络混合图神经网络的机器学习模型
图源:论文截图
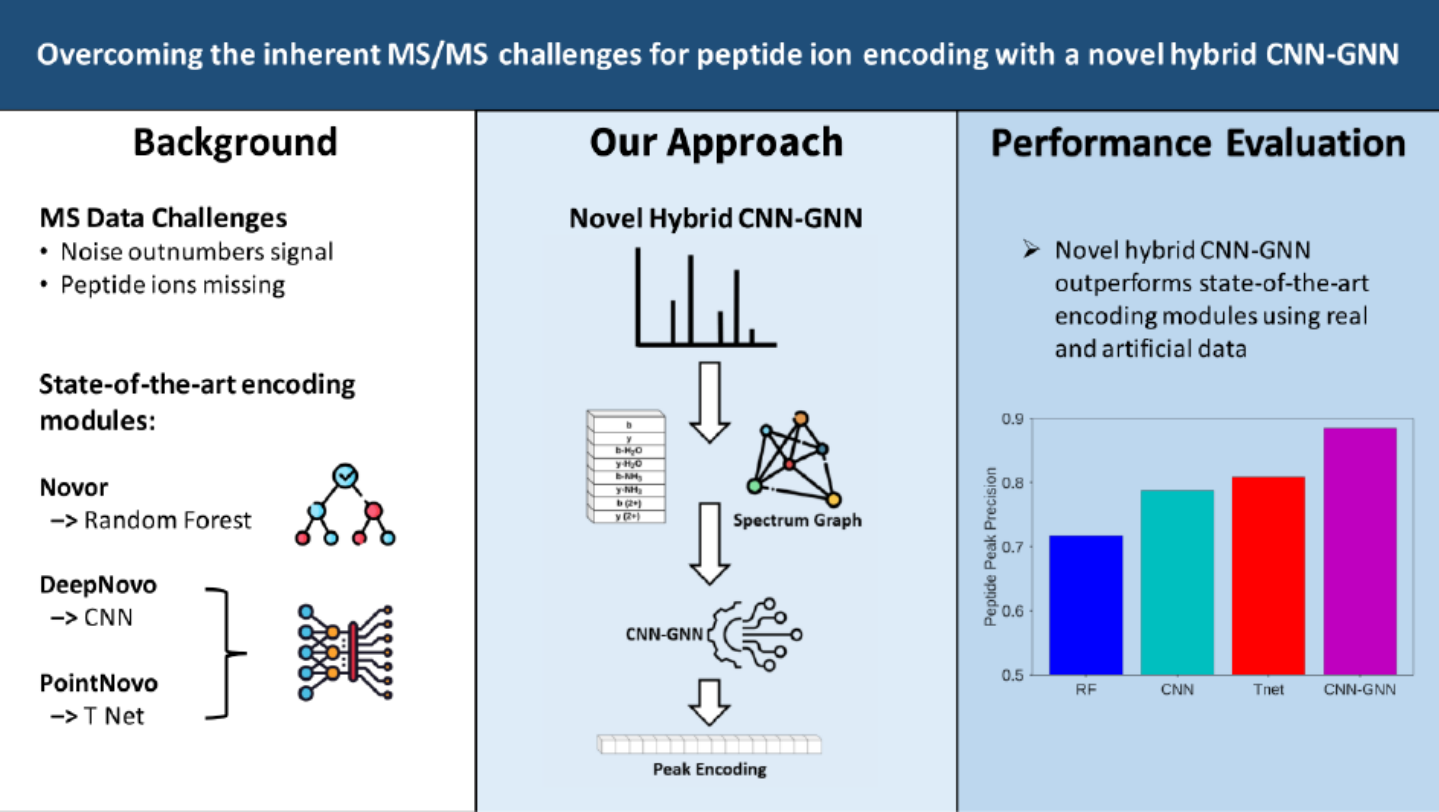
图源:文章Abstract
来自爱尔兰Galway大学的Kevin McDonnell团队提出了一种卷积神经网络混合图神经网络的机器学习模型,用于de novo肽段测序中的片段峰编码,从而在MS质谱中识别氨基酸。该团队通过比较这些模块在MS谱图中识别b、y离子的能力,将提出的编码模块与目前最先进的de nove测序算法中使用到的编码模块进行的对比。其中包括了真实数据和人工数据的综合,包括不同水平的噪声和肽峰缺失。
结果在AUC和平均精度这两个度量指标下,这项工作提出的编码模块在所有数据集中的表现最佳。
这项工作揭示目前最新算法在肽离子编码方面的局限性,所提出的模型提供了替代方案,对de nove肽识别算法的发展具有重要意义。
♦ AI揭示多组学数据与药物关联
图源:论文截图
图源:文章图片
利用多组学分析技术对临床队列进行分析有可能揭示出患者水平的疾病特征以及分析个性化的治疗反应。但如何结合各个组学(即多模态)的数据是一个具有挑战的任务。为了更好的整合多组学的数据,作者基于传统深度学习模型VAE搭建了多组学变分编码器(MOVE),并将模型应用于789名2型糖尿病患者队列,模型输入为个体水平的非组学、多组学数据以及药物状态的有无,以此来确定药物和组学的关联性。为了增强模型的可解释性,对神经网络的权重进行分析以及SHAP方法研究特征各个特征是如何影响个体在空间中的定位。进一步,作者模拟了一次扰动的特征输入方法以及新的分析方法(MOVE t-test、MOVE Bayes)来评估模型是否学习到了药物和多组学之间的关系,结果表明两种分析方法表现良好,其FDR为0.05。随后,团队应用MOVE框架识别DIRECT数据集中的药物关联,发现T2D生物标志物的变化与二甲双胍有关、二甲双胍和奥美拉唑与肠道菌群存在一定关系。同时还发现了一些药物引起组学变化之间的相似性。该研究不仅可以发现新的生物标志物,还可以从多组学数据上从患者层面进行药物选择。该研究为多组学的融合分析提供了一种可行的思路,同时,作者也提出了未来继续融合其它组学数据的期望。
https://www.nature.com/articles/s41587-022-01520-x
♦ 多模态分析方法
图源:论文截图
图源:文章图片
自2015年以来已开发30多项单细胞技术,然而数据整合面临多维度/高噪音/海量数据多方面问题。作者详述了多组学尤其是转录组与蛋白组多模态映射的相关算法及各自的优缺点(如将转录组和蛋白组进行关联映射的CiteFuse)。即便是非同一细胞来源的多组学数据分析依然在临床研究中具有重要意义(如揭示慢性肾病中肌成纤维细胞活化的机制)。作者乐观估计未来海量数据多模态联合分析将发挥更大作用。
https://www.nature.com/articles/s41581-021-00463-x
♦ 一种基于浏览器的开源 ML 工具
图源:论文截图
图源:文章Abstract
哥本哈根大学Strauss课题组开发了“OmicLearn”(http://OmicLearn.org):一种基于浏览器的开源 ML 工具,可以快速探索各种 ML 算法对实验数据集的适用性。可以在线使用或者安装在本地。他们利用 OmicLearn 来评估基于 ML 的分类将患者与对照组区分开来的程度。OmicLearn 将数据反复拆分为不同的子集,训练分类器并提供性能指标。所得 ROC 曲线的平均曲线下面积 (AUC) 为 0.90 ± 0.08,PR 曲线的平均 AUC 为 0.92 ± 0.06。支持探索性数据分析(PCA、层次聚类)、数据预处理(若干归一化算法)、特征选择(如极度随机树)、分类算法(LinearSVC、AdaBoost等)以及交叉验证算法。通过该工具可以不用编程的实现生物标记物的发现。
♦ Science首席Editor对可以自动写文章问答系统ChatGPT的评论
图源:文章截图
Science 的首席Editor H. Holden Thorp本周评论了关于最近很火的可以自动写文章问答系统的ChatGPT ,强调Sceince系列期刊是不接受使用AI工具如ChatGPT写的文章的:这种基于AI自动生成文章因为产生自机器而不能证明“该作品是原创的”,相当于一种抄袭的。他们目前正在改Editorial Policies, 加一个rules表明science杂志不能接收使用由 ChatGPT(或任何其他人工智能工具)生成的文本,数字、图像或图形文章,反这些政策将将构成科学不端行为, 他们认为这和与篡改图像或抄袭现有作品无异。AI机器的确发挥着重要作用,但它只是人们提出假设、设计实验方法和结讨论果的工具。最终的科研著作必须来自我们头脑中的“计算机”来表达。
笔者认为他们对AI抄袭的定义其实是对AI机器没有真人的贡献。我们可以采取中和的方式,一部分AI一部分自己写,根据我们自己的文章生成一个对话系统写初稿,然后再结合自己的实验结果修改,这种文章,至少还是避免了一些大框架逻辑的错误和语言的错误,还是提高了一部分效率。
https://www.science.org/doi/10.1126/science.adg7879
评论:张芳菲
♦ Cyclic Ion Mobility-Mass Spectrometry技术
图源:论文1截图
图源:论文2截图
图源:文章Abstract
第一篇文章介绍了Cyclic Ion Mobility-Mass Spectrometry技术,在传统的IM-Q-TOF仪器上,用98cm长的的圆形行波离子淌度池替代线性离子淌度池,通过循环装置扩展了离子淌度分离的优势。数据显示,通过3次循环分离,5种寡糖(拥有同分异构体的前体离子和碎片离子)可以很好地区分,显示了其在同分异构体的区分上有非常大的潜力。
第二篇文章介绍了基于该技术成功分离了阿尔兹海默症病人的脑组织样本中tau蛋白的异构磷酸肽,这对神经性退行性疾病的病理性蛋白修饰的研究提供了一个可行性的分析流程。
♦ 基于分子互作网络和开源数据库建立疾病特异性的药物蛋白连接谱
图源:论文截图
图源:文章Figure
随着基因-药物的分子连接图谱揭示越来越多的基因和药物化合物之间的生物计量关系,如何针对特定疾病提供具有临床应用价值的药物化合物信息,2009年时产生了一次针对疾病-蛋白-药物连接图整合的计算尝试,在没有生成蛋白质-基因-药物化合物连接图的年代,通过整合
1)蛋白相互作用网络,
2)蛋白-基因名检索及同义词扩展,匹配pubmed摘要中提到的药物名称
3)生成疾病相关蛋白与药物化合物矩阵,在此基础上进行过滤和聚类分析。该文章以AD为例子,展示了一个针对AD的计算分子(蛋白-药物)连接图,并筛选出地尔硫卓和奎尼丁肯可能成为作为AD治疗的候选药物。截至目前,该文章共引用两百余次,但很遗憾的是,跟据该方法搭建的支持搜索疾病相关的蛋白-药物关系的网站已经不再维护。结合GUOmics的研究,可以推广到,如果我们拥有实验得到的蛋白-药物连接图,可以整合CORUM,Bioplex等通过实验得到的complex resource,通过in silico得到蛋白质复合物-蛋白-药物化合物之间的连接图。
♦ 一种卷积神经网络混合图神经网络的机器学习模型
图源:论文截图
图源:文章Abstract
来自爱尔兰Galway大学的Kevin McDonnell团队提出了一种卷积神经网络混合图神经网络的机器学习模型,用于de novo肽段测序中的片段峰编码,从而在MS质谱中识别氨基酸。该团队通过比较这些模块在MS谱图中识别b、y离子的能力,将提出的编码模块与目前最先进的de nove测序算法中使用到的编码模块进行的对比。其中包括了真实数据和人工数据的综合,包括不同水平的噪声和肽峰缺失。
结果在AUC和平均精度这两个度量指标下,这项工作提出的编码模块在所有数据集中的表现最佳。
这项工作揭示目前最新算法在肽离子编码方面的局限性,所提出的模型提供了替代方案,对de nove肽识别算法的发展具有重要意义。