♦ 使用多模态深度学习预测AD进展的方法

图源:文章截图
来自宾夕法尼亚州立大学的团队于scientific reports发表了使用多模态深度学习预测AD进展的方法:阿尔兹海默症(AD)是一种不可逆的神经退行性疾病,其特征是大脑中淀粉样斑块和神经元纤维纠缠的异常积累,如今任何方法都无法治愈AD。
早期检测AD对于及时治疗以及延缓其进程十分有帮助。轻度认知障碍(MCI)这个概念就此被提出。有些MCI患者后期会变为AD患者,有些则不会,研究团队提出了一个综合框架,可结合影像学生物标志物、脑脊髓液标志物以及认知表现生物标志物,能够准确预测出MCI患者是否发展为AD患者(ACC=81%,AUC=0.86)。
具体方法为使用RNN预测基线开始后Δt时间后的转化结果。同时作者还比较了单模态以及混合多个模态的预测结果,结果表明,混合使用多个模态比使用单模态能取得更好的效果。如今深度学习发展迅速,对于时序数据处理方面,transformer模型已经基本取代了RNN,transformer预测AD也许是一个可行的方法。
https://www.nature.com/articles/s41598-018-37769-z
♦ 再利用的药物可能与 AD 发病率的降低有关

图源:论文截图
图源:文章Graphical abstract
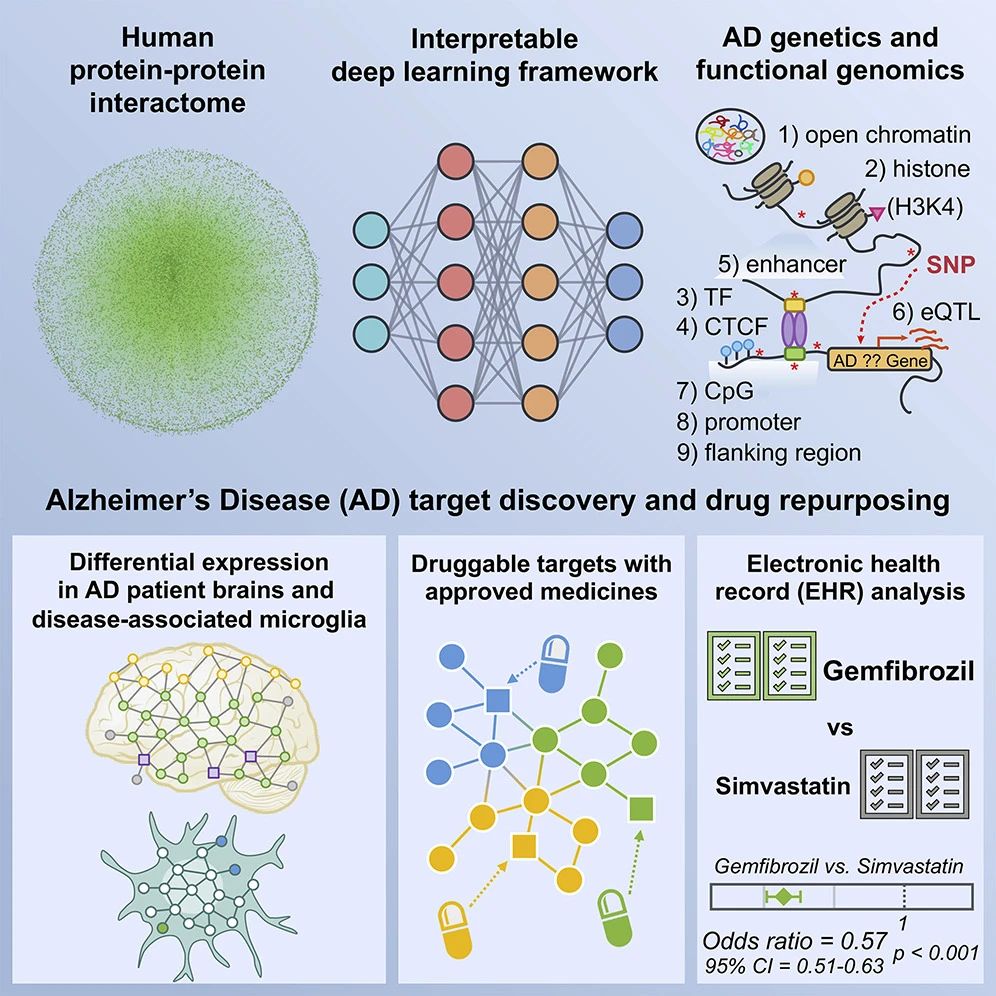
来自美国克利夫兰医院Feixiong Cheng团队在Cell Reports的一项工作,使用发表了的GWAS 、多组学数据、临床数据结合基于网络的、解释深度学习(NETTAG)的模型,对四种药物进行药物再利用评估针对阿尔茨海默病的治疗。
具体地,他们使用了人蛋白互作组数据,表观遗传组数据如TF, CTCF组蛋白,CpG,和GWAS的eQTL数据先组成神经网络的图先预测出156个AD风险基因,然后再结合多组学的差异表达数据(包含六个蛋白质组老鼠AD研究)、FDA获批的药物靶点数据库对预测的结果进行进一步评估,最后通过临床病人的预后数据中进行验证筛选出了四个可以再利用的药物(布洛芬、吉非贝齐、胆钙化醇和头孢曲松)是可能与 AD 发病率的降低有关。
https://linkinghub.elsevier.com/retrieve/pii/S2211124722015959
♦ 使用PET脑图为阿尔茨海默病的早期预测开发的深度学习算法

图源:论文截图
Yiming Ding来自加州大学旧金山分校的放射学和生物医学成像系,在2018年开发和验证一种深度学习算法,该算法可预测大脑的氟 18 (18F) 氟脱氧葡萄糖 (FDG) PET 最终诊断为阿尔茨海默病 (AD)、轻度认知障碍或两者都不是,并将其性能与放射读取器的性能进行比较。
数据集为1002名AD患者的2019张PET脑图像,独立测试集为40名AD患者的40张PET脑图像。90%用来训练,10%和独立测试集用来测试。训练模型为InceptionV3是谷歌Inception系列里面的第三代模型。使用灵敏度、特异性、ROC、显著图和 t 分布随机邻域嵌入对模型进行分析。
最终结果表明通过使用PET脑图,为阿尔茨海默病的早期预测开发的深度学习算法在 100% 的灵敏度下达到了 82% 的特异性,平均比最终诊断早 75.8个月。
♦ directLFQ和Speedy-PASEF:更快提升质谱蛋白质组的通量
图源:论文截图

图源:论文截图
Matthias Mann和Markus Ralser背靠背在BioRxiv更新了他们最新的研究进展directLFQ和Speedy-PASEF,都是为了更快提升质谱蛋白质组的通量。
directLFQ 是一个提升大规模数据分析速度的方法,它通过在对数空间对齐样本和离子轨迹约化来计算蛋白的定量,一个重要特点是可以线性的约化大样本量的文件,特别适用于大规模队列数据的研究。
速度结果上他们展示了一个10 min的时间内鉴定超过1万的蛋白,在集群上2小时内可以实现对10万个蛋白质组质谱文件的定量,并且展示了在Meier et al.的基线Mixed Orbitrap LFQ数据中超过MaxLFQ在DDA和在Huang et al. 的Obtritrap DIA的效果,应用数据为细胞器蛋白质组学数据也展示了和maxLFQ类似的生物学结论。这个方法也可以结合到他们组目前的AlphaPept体系里也可以独立作为python包或者GUI安装。https://github.com/MannLabs/dire
Speed-PASEF 使用了分析流对PASEF加上最新版本DIA-NN 1.8.1,3 μg K562细胞5 min梯度单针5844, 7%CV中值, 微流在5.6 min鉴定到5286个蛋白。液相为800 μl/min Agilent1290加LunaOMEGA 柱子30x2.1 mm短柱子,质谱为TimsTOFPro。
首先方法优化上他们先使用5 min和正常优化了9个进样量10 ng-3000 ng,进行定性数量评估和定量CV和混合物种定量值分布评估,应用仍旧选择了血浆新冠蛋白质组队列10名健康和30名非轻症患者采集了398个3 min队列样本,生物学角度验证了之前发现的一系列差异蛋白(ALB,SAA1, CRP, C1S)。这项工作验证了分析流液相结合diaPASEF和DIANN分析的可能性,利用了TIMS了某种程上度克服了分析流需要的大进样量的问题让分析流液相可以拓展到100ng以下的应用。
♦ 常规组织学和空间蛋白质组学中膜载玻片的标准化和可重复工作流程

图源:论文截图
德国马克斯·普朗克生化研究所Matthias Mann团队于2023年02月20日在BioRxiv上发表了“常规组织学和空间蛋白质组学中膜载玻片的标准化和可重复工作流程”的预印本论文。
这项工作在其去年在Nature Biotechnology上发表的“Deep Visual Proteomics defines single-cell identity and heterogeneity” (https://doi.org/10.1038/s41587-022-01302-5)的文章基础上进行了方法学上的优化,解决了组织水平的空间蛋白质组学领域一个很重要的问题,即免疫染色,因为抗原修复的效果会决定染色的成败,但是通常这个过程是很剧烈的,比如说需要高热与高压的条件等,而基于激光显微切割(LMD)的过程需要在膜片上进行取样,但常规使用的膜片比较脆弱,所以他们将金属框的膜片换成了载玻片形制的膜片,之后通过甘油孵育解决了覆盖的塑料膜不平整从而影响成像焦平面的问题,并通过与Leica的LMD系统相结合,提高了免疫染色、成像与取样的成功率。
他们同时也表明甘油与基于质谱的蛋白质组学完全兼容,其添加不会影响蛋白质组的鉴定深度和重复性。该工作的应用为针对人体FFPE皮肤样本中相邻的基底上和基底角质形成细胞的比较分析,共鉴定到超过3500个蛋白质 ,且较好地保留了不同类型细胞的蛋白质空间表达的信息,所使用的质谱系统为Evosep One 整合timsTOF SCP。
此项工作的完成,让Matthias Mann团队所开发的Deep Visual Proteomics更具有兼容性和应用性,最终助力于临床蛋白质组学的研究。
♦ 共价标记与质谱联用

图源:论文截图
共价标记(CL)与质谱联用可用作分析蛋白质-蛋白质复合物的结构性质。但是从实验中获取到的数据是稀缺的,并不能明确地阐述蛋白质的结构。AlphaFold2是DeepMind公司提出的用于预测单蛋白质分子结构的模型,可达到原子精度,但在蛋白质复合物预测层面有一定的缺陷。
蛋白质结构领域大牛David Baker组开发的RosettaDock可以结合共价标记数据,来进行蛋白质复合结构的预测。在这项研究中作者首先利用AlphaFold2模型预测蛋白质的三维结构,随后利用RosettaDock模型结合CL数据预测蛋白质的复合结构。
在纯计算模拟(AlphaFold2 + RosettaDock)的情况下,TM-socre为0.70,DockQ为0.21,结合CL实验数据后,两个分数分别提升为0.84和0.50。实验与最先进的计算工具相结合,显著提高了蛋白质复合物预测的准确度。作者团队还提出了探究使用其它类型的标记方法与质谱联用,继续搭建更加复杂的预测工具。