







2023年8月14日,在美国杨百翰大学、圣路易斯华盛顿大学、麻省理工学院和哈佛大学布罗德研究所、美国梅耶癌症中心等机构领导下,CPTAC(美国临床蛋白质组学肿瘤分析联盟)在 Cell、Cancer Cell 共发表了4篇关于癌症研究的论文(3篇article和1篇perspective),共同强调了多组学方法在深入理解癌症发生机制、疾病特征以及潜在治疗途径方面的重要性。它们提供了全面的数据资源,有助于推动癌症研究的前沿。
1 Proteogenomic data and resources for pan-cancer analysis

通过肿瘤基因组图谱,可以深入了解驱动肿瘤发生的突变。然而,这些图谱主要关注基因组,对蛋白质及其修饰的数据较为缺乏。为了将基因型与表型相连接,作者提出了用于泛癌分析的 “蛋白质基因组学”(proteogenomics)。该研究从蛋白质组学和基因组学的角度对肿瘤进行分析,将基因组异常与癌症表型相连接,为泛癌研究提供了丰富的多组学数据。
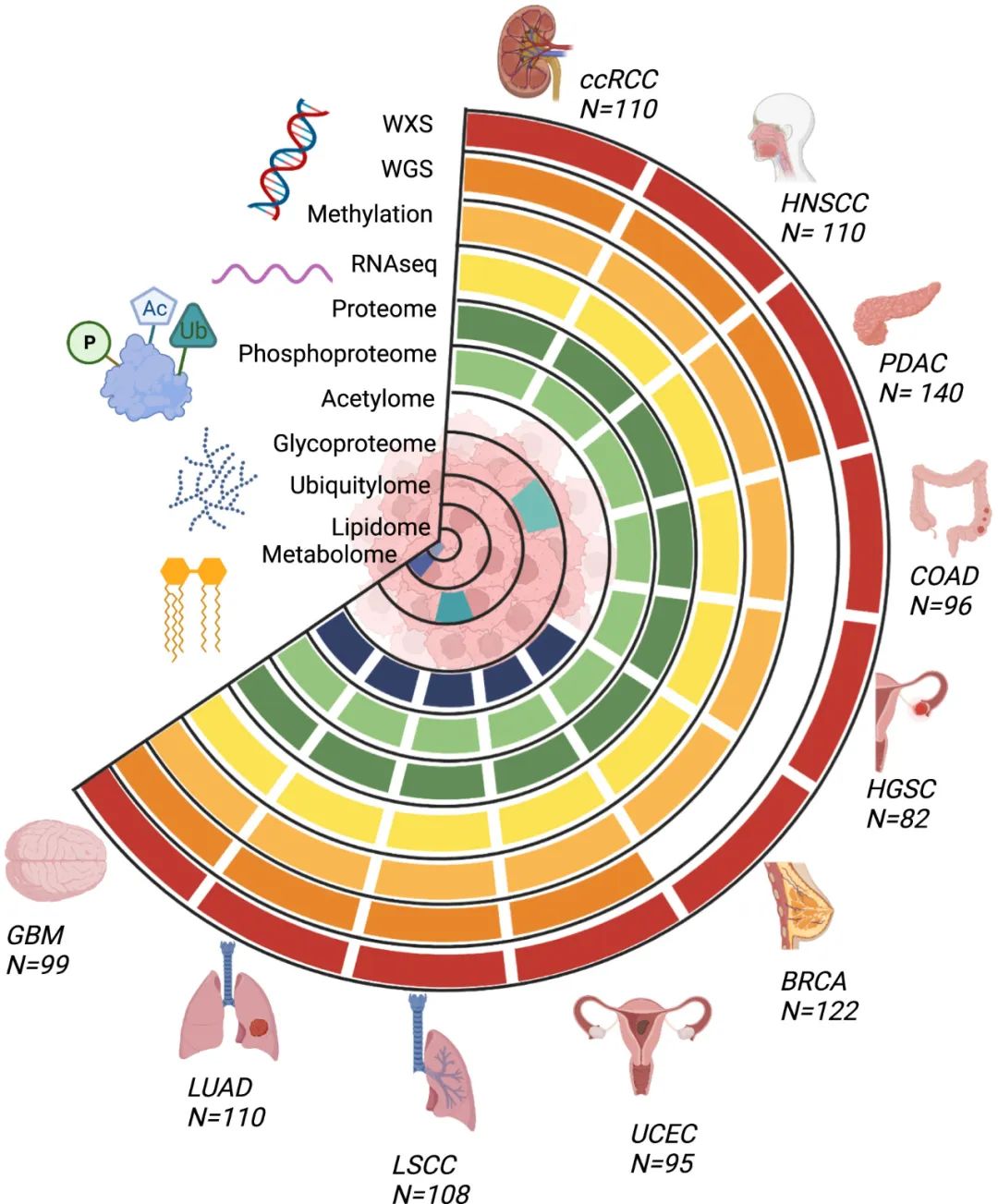
CPTAC泛癌数据集的肿瘤类型和数据类型
为了促进泛癌研究,研究团队生成了10个队列中的1000多个肿瘤的基因组、转录组、蛋白质组和临床数据,以建立一个有力的数据集用于科学发现。
该研究还介绍了CPTAC全癌症工作组在数据协调、数据传播和为促进生物学发现提供计算资源方面的努力,以帮助生物学发现。研究还讨论了多组学数据整合和分析的挑战,特别是在处理核苷酸测序和质谱蛋白质组学数据时所面临的独特挑战。同时,该文提供了一个CPTAC的数据资源,旨在为广大癌症研究界的人员提供有关数据处理、分析和解释的方法和思路。
论文链接:
https://www.sciencedirect.com/science/article/pii/S1535610823002192?via%3Dihub#bib3
2 Integrative multi-omic cancer profiling reveals DNA methylation patterns associated with therapeutic vulnerability and cell-of-origin

DNA甲基化在建立和维持细胞身份方面发挥着关键作用,但在肿瘤发展过程中经常失调,并与其他遗传变异密切相关。文章强调了整合多组学数据的重要性,以全面了解肿瘤特异性甲基化如何影响转录和翻译。通过使用蛋白质组学数据作为生物活性的直接测量,研究人员旨在发现DNA甲基化驱动基因,并了解它们在肿瘤发展中的作用。
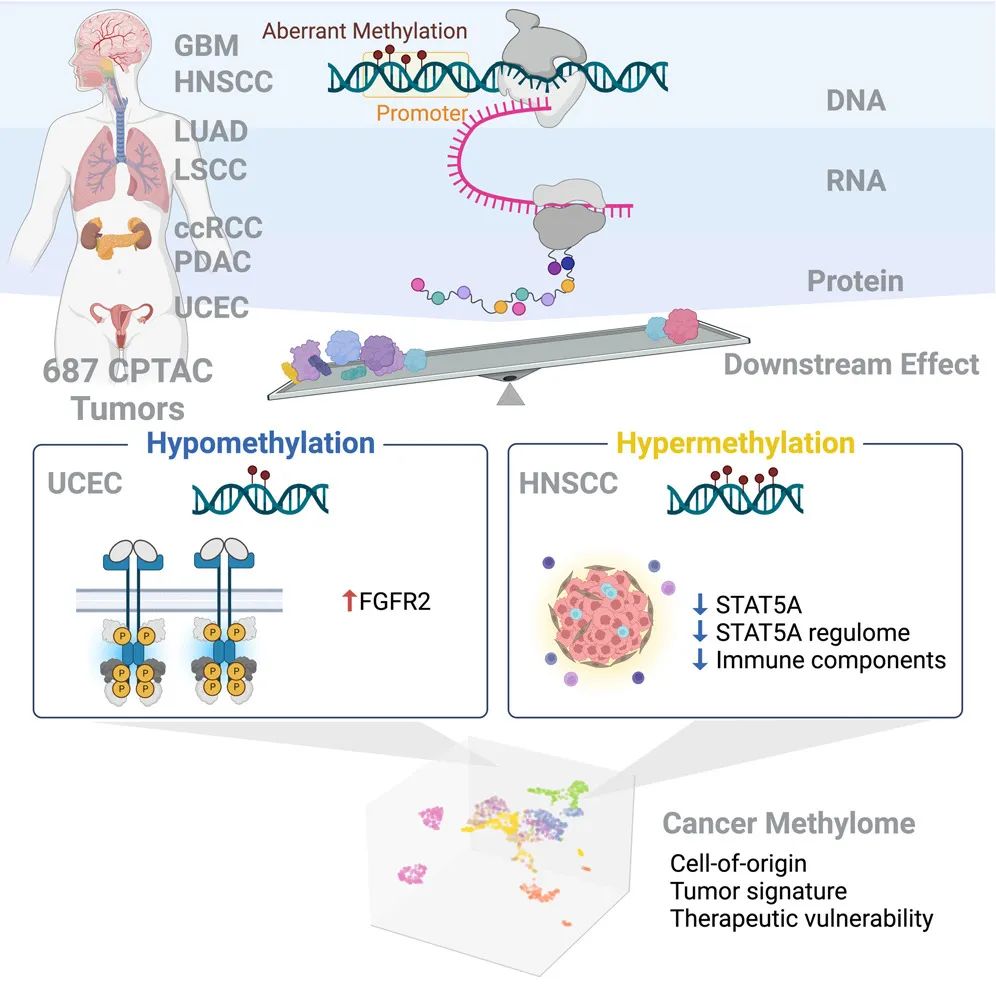
该研究通过对来自肾脏、脑、胰腺、肺、头颈部和子宫内膜中的687个肿瘤及其匹配的非浸润癌旁组织(non-involved adjacent tissues)进行多组学分析,识别与RNA和蛋白质丰度变化相关的异常甲基化,揭示了DNA甲基化在细胞身份建立和维持中的关键作用,并构建了一个泛癌目录。
研究发现了与特定细胞系相关的表观遗传驱动因子,例如子宫内膜癌中低甲基化的FGFR2。研究还表明高甲基化的STAT5A与广泛的调控子下调和免疫细胞耗竭相关,暗示STAT5A表达的表观遗传调控构成了鳞状瘤肿瘤免疫抑制的分子开关。
此外,研究还证明甲基化亚型富集信息可以解释细胞起源、肿瘤内异质性和肿瘤表型。总体而言,研究识别了驱动转录和翻译变化的顺反式DNA甲基化事件,揭示了肿瘤的表观遗传景观及其细胞起源的作用,为我们深入理解肿瘤的表观遗传特征以及其治疗策略提供了新的视角。
论文链接:
https://www.cell.com/cancer-cell/fulltext/S1535-6108(23)00253-2#%20
3 Pan-cancer proteogenomics connects oncogenic drivers to functional states

癌症驱动因子是指推动癌症发生的关键基因异常,然而,我们仍未完全理解它们的确切分子机制。该研究通过对来自十种不同癌症类型的数据进行分析,展示了不同癌症类型之间的多组学特征,并根据这些特征将肿瘤样本聚类为不同的子群。
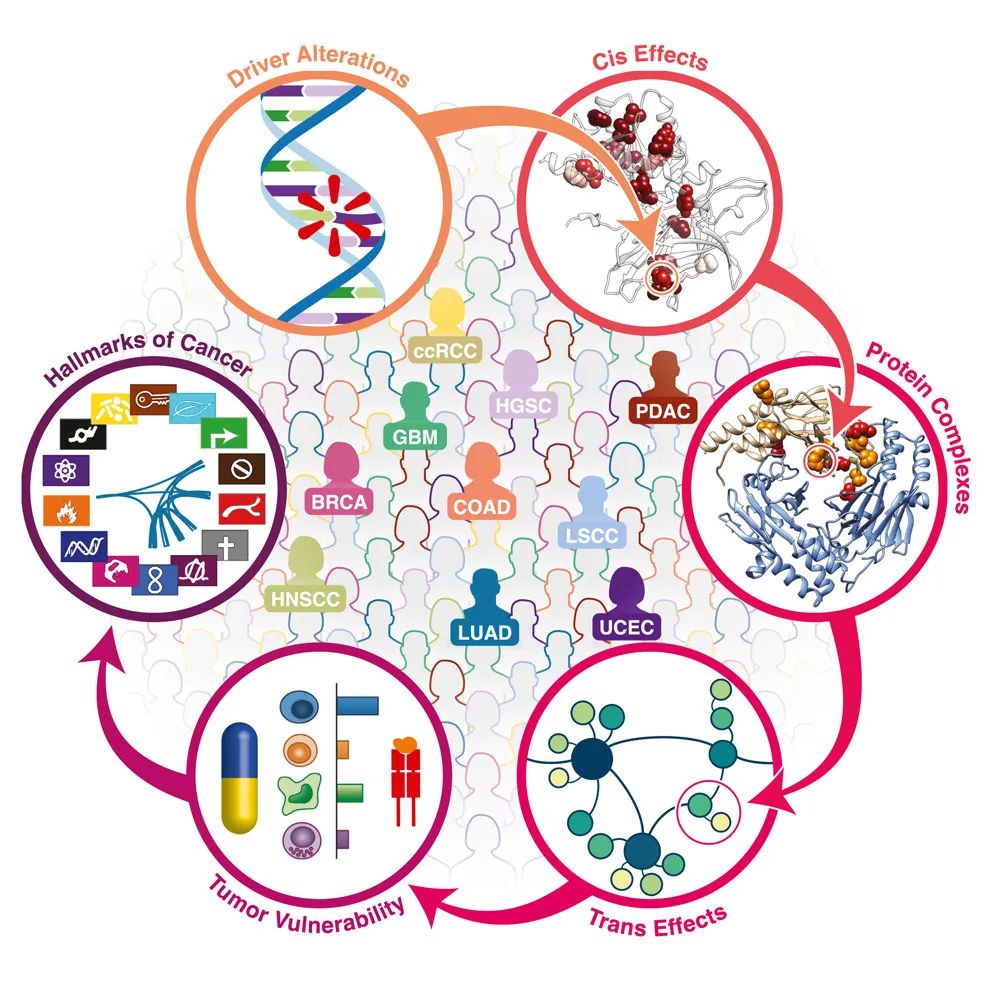
作者关注了肿瘤样本中癌症驱动基因的突变如何影响其RNA、蛋白质和磷酸蛋白水平,揭示了突变和拷贝数变化与蛋白质相互作用网络的重构之间的关联,特别是大多数癌症基因汇集于类似的分子状态,这些状态由基于序列的激酶活性谱所表示。
此外,文章强调了免疫治疗的潜在价值。预测的新抗原负荷与测量的T细胞浸润之间的关联表明了免疫疗法的潜在易感性。另外,作者还研究了癌症标志物的模式,发现其与多基因蛋白质丰度的变化有关,不同的癌症类型显示出不同的标志物模式。最后,作者通过蛋白质共变分析,研究了肿瘤样本中基因突变对蛋白质相互作用的影响,以及肿瘤基因的distal trans-effects。
总的来说,这项工作展示了全蛋白质组学在理解致癌驱动因子的功能状态以及它们与癌症发展的联系方面的价值,克服了单一癌症类型研究的局限性,这对于进一步的癌症研究和临床应用具有重要意义。
论文链接:
https://www.cell.com/cell/fulltext/S0092-8674(23)00780-8#%20
4 Pan-cancer analysis of post-translational modifications reveals shared patterns of protein regulation

翻译后修饰(PTMs)在调控正常和癌症细胞的细胞信号和生理过程中起着关键作用,质谱技术的发展使得可以高通量、高精度、高灵敏度地测量PTM水平,从而更好地了解它们在标志性癌症过程中的作用、普遍性和相互影响。
之前的跨癌基因组研究已经证明,调查不同癌症类型之间的常见基因和通路变化,有助于我们理解驱动癌症的基本分子事件。本文探讨了PTMs在正常细胞和癌细胞中调节细胞信号传导和生理功能的关键作用。
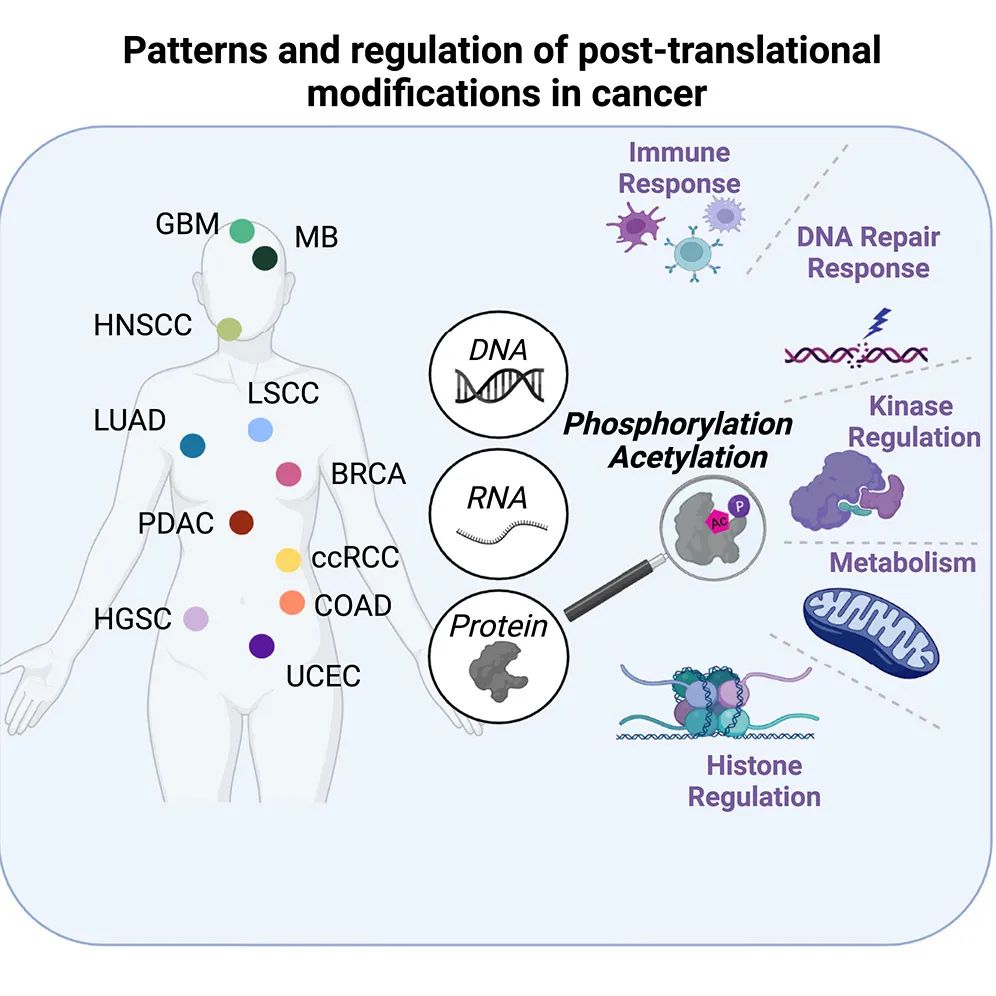
该研究分析了来自1110名患者的11种癌症类型的最大规模的蛋白质组学数据集,包括PTM数据(其中10种来自CPTAC)。研究揭示了涉及标志性癌症过程的蛋白乙酰化和磷酸化变化的全癌症模式。这些模式揭示了来自不同癌症类型的亚组,包括那些由磷酸化驱动的DNA修复失调、由乙酰化驱动的免疫应答相关的代谢调节改变、由乙酰化和磷酸化之间的串扰引起的受损激酶特异性,以及改变的组蛋白调控。
文章的结果部分描述了跨癌症数据集的概述,包括样本数量、肿瘤类型、基因表达、蛋白质水平和PTM水平等方面的信息。他们使用SignatureAnalyzer等分析工具对数据进行了处理和分析,揭示了33个跨癌症多组学特征。他们对不同癌症类型中的共享和差异性生物学进行了深入的研究,包括DNA修复缺陷、免疫响应、代谢调节和PTM之间的相互作用等。文章还详细讨论了特定的研究发现,如DNA修复缺陷肿瘤中的磷酸化模式、代谢亚型中的乙酰化模式以及PTM之间的预测相互作用等。
该研究也存在一定的局限性,包括样本数量和样本类型的限制,以及质谱分析的假阴性率等。论文作者认为,未来,更大规模研究的开展将有助于更全面地了解癌症中的蛋白质组学机制。
总体而言,这项研究通过对大规模的跨癌症蛋白质组学数据进行综合分析,凸显了PTM调控的丰富生物学,并揭示了潜在的新治疗途径。这些发现对于揭示癌症的基本机制、寻找新的治疗靶点以及开展个体化治疗具有重要意义。
论文链接:
https://www.cell.com/cell/fulltext/S0092-8674(23)00781-X?_returnURL=https%3A%2F%2Flinkinghub.elsevier.com%2Fretrieve%2Fpii%2FS009286742300781X%3Fshowall%3Dtrue#%20
CPATC
2006年年初,美国国家癌症研究所(National Cancer Institute,NCI)开始了一项为期5年、耗资上亿美元的工作:建设临床蛋白质组肿瘤分析联盟(Clinical Proteomic Tumor Analysis Consortium,CPTAC)。
2011年,CPATC正式推出,旨在通过应用大规模蛋白质组和基因组分析或蛋白质基因组学来加速对癌症分子基础的了解。当时,CPATC率先对结直肠癌、乳腺癌和卵巢癌进行蛋白质组学综合分析,以揭示对这些癌症类型的新见解,例如识别以蛋白质组为中心的亚型、通过拷贝数变化和蛋白质丰度的相关分析确定驱动突变的优先级、并通过翻译后修饰了解癌症相关途径。
截至目前,CPTAC 向基因组数据共享中心 (GDC) 提供了共 1500 多名癌症患者的基因组数据(癌症类型包括子宫内膜癌、肾癌、肺腺癌和鳞状细胞癌、乳腺癌、结肠癌、卵巢癌、脑癌、头颈癌和胰腺癌)。


