






3月6日,伦敦MRC医学科学研究所(LMS)和慕尼黑亥姆霍兹中心功能表观遗传学研究所的学者在 Nature 发表了新的文章 Decoding chromatin states by proteomic profiling of nucleosome readers。

图1 论文截图
文章通过系统性的蛋白质组学分析,揭示了核小体 “阅读器”(nucleosome readers)蛋白质如何解读复杂的染色质状态,从而为染色质调控的机制提供了重要洞察。
染色质(chromatin)由DNA和组蛋白(histone)的修饰组合而成,这些修饰形成特定的模式,用于标记基因组的功能区域,如启动子、增强子和异染色质等。许多核蛋白能够识别单个修饰,但对于复合修饰模式的解读仍有待研究。
在该研究中,研究人员通过多维度的蛋白质组学策略,系统研究了近2000种核蛋白质与80多种代表启动子、增强子和异染色质状态的修饰双核小体之间的相互作用。
结果1:
染色质阅读器的蛋白质组学分析
研究人员通过SILAC*核小体亲和纯化(SILAC nucleosome affinity purification,SNAP)技术,系统性地分析了核小体修饰的相互作用网络。他们设计了一系列含有特定修饰的核小体,通过正反向SILAC核小体亲和纯化实验来探索修饰对蛋白质招募(recruitment)或排斥(exclusion)的影响。
研究人员进行了大规模比较不同染色质状态的相互作用组学研究,涵盖了单个核小体上的多种修饰模式,如甲基化、乙酰化和组蛋白变异等。通过这些实验,他们得到了近2000个与不同染色质状态相互作用的核蛋白,为后续对染色质状态解读提供了重要数据支持。
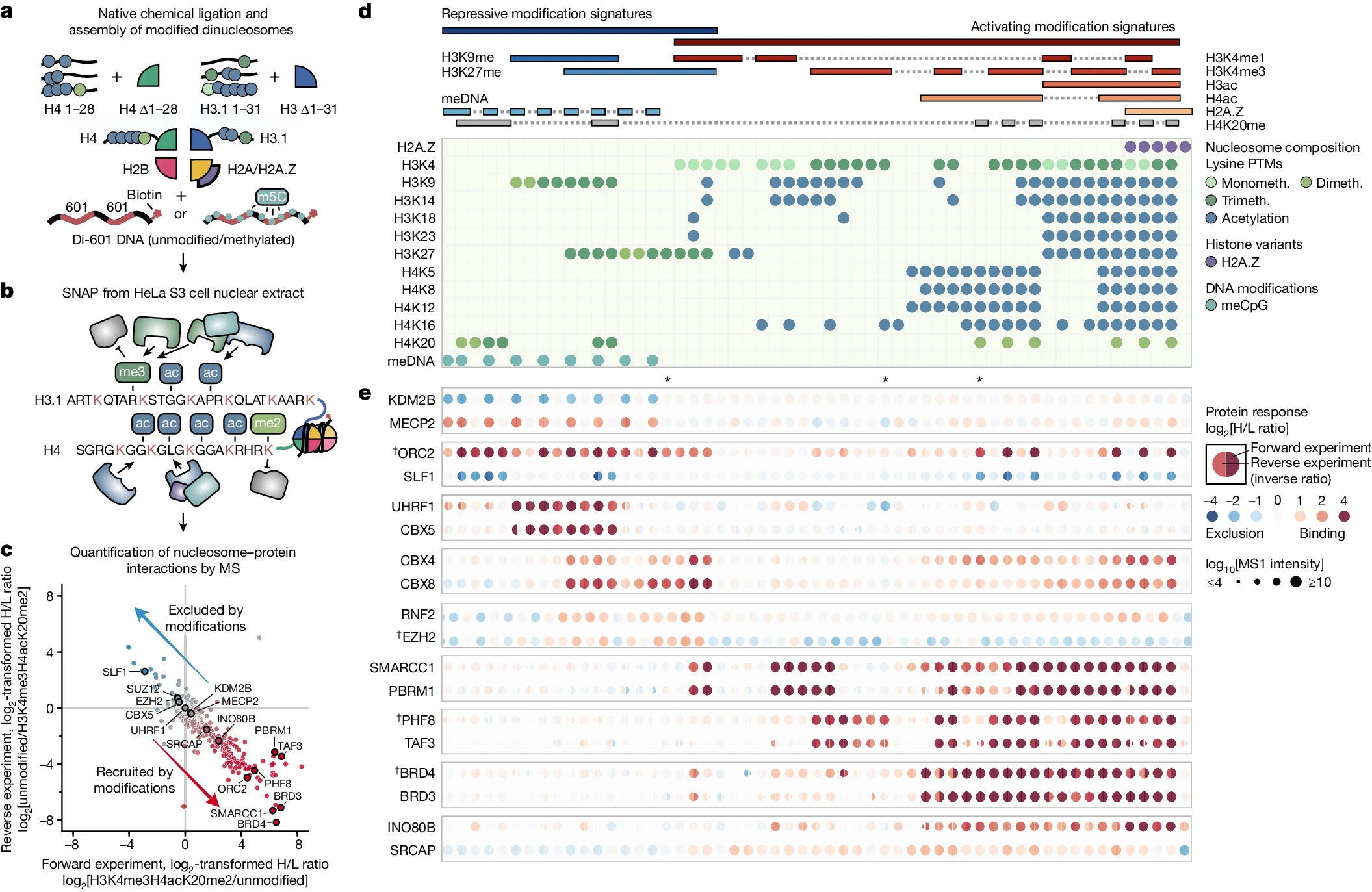
图2 通过SILAC双核小体亲和纯化大规模鉴定染色质阅读器
*注:SILAC,细胞培养条件下稳定同位素标记技术(Stable isotope labeling with amino acids in cell culture),是基于质谱的定量蛋白质组学中一种简单而稳定的方法。
结果2:
MARCS系统绘制染色质结合反应
研究人员通过比较不同修饰状态下蛋白质的相对招募或排斥程度,发现了许多核蛋白的特定核小体结合模式。他们开发了一个染色质状态调控修饰图谱在线资源MARCS(Modification Atlas of Regulation by Chromatin States),用于交互式地可视化这些核小体相互作用的数据。
利用MARCS,研究人员展示了核蛋白质对激活和抑制修饰状态的广泛响应,揭示了核蛋白与不同染色质状态之间的相互作用模式,包括对单一修饰的简单响应(例如MECP2的招募或KDM2B被DNA甲基化所排斥)和对多重修饰的复杂相互作用(如起始识别复合物(ORC)对H3K9、H3K27或H4K20甲基化的招募,并进一步受到DNA甲基化的刺激)。此外,他们还发现了许多新的染色质与核蛋白相互作用,拓展了对染色质状态解读的认识。
结果3:
无偏差预测结合特征
通过分析核蛋白对不同修饰特征的响应,研究人员发现许多核蛋白表现出复杂的结合模式,无法仅通过单一特征解释。为了描述这种复杂的结合行为,他们将核小体结合谱分解成驱动这些结合的单个核小体特征,并通过统计学方法计算了每个核蛋白对于不同修饰特征的相对结合程度。
结果显示,许多核蛋白对多个修饰特征均有响应,并且这些响应可以归纳为40种主要结合模式。这种方式使得他们能够量化描述数百种蛋白质的染色质结合行为,并将复杂的结合剖析成一组关键特征,这些特征要么正向,要么负向地调节它们与修饰的核小体的结合。
此外,研究人员通过整合公共的ENCODE ChIP-seq数据验证了这些修饰特征对核蛋白的结合行为的准确性。这些结果为进一步理解核蛋白质如何解读不同染色质状态提供了重要线索,有助于揭示染色质状态调控的机制。
结果4:
缺乏独特的H3K4me1“阅读器”
研究人员通过特征效应分析发现,蛋白质对H3K4甲基化的结合存在差异。对于启动子标记H3K4me3,他们发现了45个被强烈招募的蛋白质,其中包括已知的H3K4me3 readers(如TFIID和PHF8),以及 31个被强烈排斥的蛋白质(如多梳蛋白抑制复合物2(PRC2))。
相比之下,增强子标记H3K4me1仅富集了一个蛋白质(BRPF3)。与这些研究结果相一致的是,研究人员的整合ChIP-seq数据分析未发现与H3K4me1强烈关联的蛋白质,而许多蛋白质更倾向于H3K4me3标记的基因组区域。这一发现得到H3K4me1和H3K4me3富集的单核小体的支持。
尽管许多蛋白质在H3K4me1和H3K4me3的ChIP中都显著富集,但其中绝大多数蛋白质更倾向于与H3K4me3修饰的染色质结合(而不是H3K4me1)。这表明了缺乏独特的H3K4me1互作网络,支持H3K4me1不是蛋白质招募至增强染色质状态的主要驱动因素的观点。
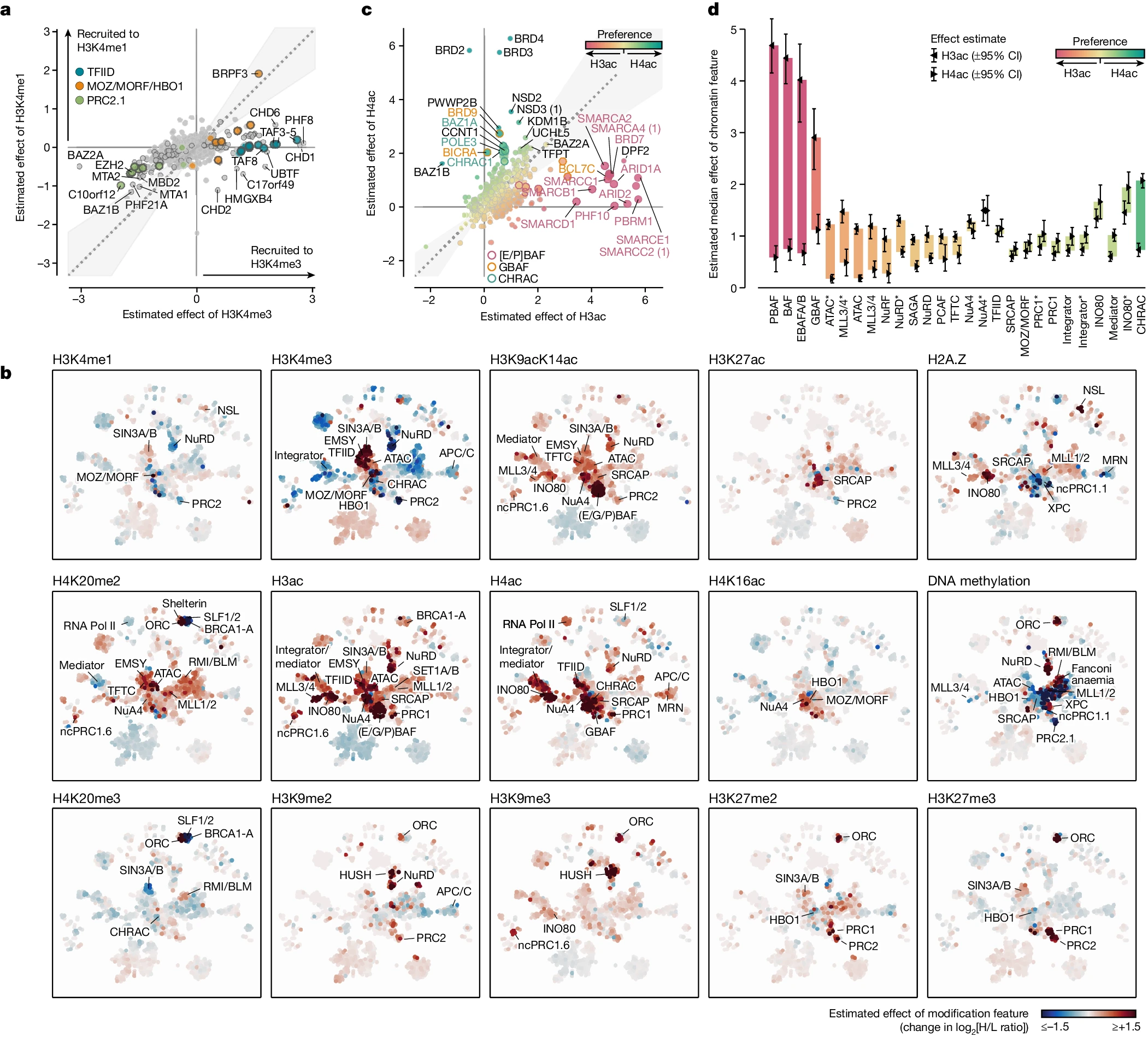
图3 蛋白质与H3K4甲基化和H3/H4乙酰化状态的差异结合
结果5:
MARCS构建蛋白质相互作用网络
通过对蛋白质复合物结合分析,研究人员发现其亚基表现出高度相似的结合行为,强调了它们的原始组成在亲和纯化中保持不变。研究人员利用这些信息重构了由相似染色质状态共同调节的蛋白质网络,并用此来预测蛋白质-蛋白质间相互作用。在此过程中,他们对几种网络推断算法进行了训练和测试,结果显示,CLR算法表现最佳。
在生成的网络中,关键的染色质调控复合物形成了聚类,随着严格程度的增加,这些聚类被解析为单独的复合物和高可信度的二元交互作用。重要的是,在综合ChIP-seq分析中,蛋白质对之间的归一化互信息估计随着预测相互作用的置信度增加而增加,表明CLR预测的网络在体内染色质相互作用中的正确性。
研究人员利用识别的局部蛋白质相互作用在MARCS中实现了相似性预测,并结合来自其他资源的蛋白质复合物信息。最后,通过将染色质特征效应估计叠加到网络上,研究人员揭示了染色质修饰反应如何驱动它们排列成紧密的子网络。
结果6:
核小体修饰和连接DNA的独立作用
除了共价修饰外,染色质状态的特征还包括连接DNA(linker DNA)的长度。研究人员进行了一系列的亲和纯化实验,利用含有不同连接DNA的双核小体来研究连接DNA对核蛋白识别染色质的影响。结果发现连接DNA长度的变化通常不会影响到染色质状态的读取,强调了MARCS中捕获的蛋白质结合响应的稳健性。
此外,连接DNA的变化也未明显影响到特异性转录因子对DNA序列的识别。总体而言,这些结果表明核小体修饰和连接DNA连接在招募蛋白质到染色质中起着相对独立的作用,强调了核小体间距对染色质结合的调控潜力。
结果7:
INO80的多价染色质结合
研究人员结合了多种分析方法,发现了ATP依赖的核小体重塑酶INO80与许多其他蛋白质的相互作用,其中包括转化生长因子β调节因子1(TBRG1)。作者发现INO80复合物对DNA连接的变化不敏感,但对多价核小体修饰特征的结合具有显著影响,尤其是对H3和H4末端尾部的乙酰化以及组蛋白变体H2A.Z的修饰。
这些结果验证了研究人员的分析和预测的可靠性,强调了研究数据的价值,并突显了这些数据用于识别先前未描述的蛋白质相互作用和复杂结合事件的价值。
总 结
综上所述,这篇文献提供了对染色质状态及其解读的全面认识。
通过将复杂的核小体结合谱分解成共同调控蛋白质和驱动蛋白质招募或排斥的不同核小体特征网络,研究人员全面展示了染色质状态是如何被核蛋白解读的,并呼吁其他研究者们利用MARCS资源深入探索数据,以推动未来染色质研究的发展。
MARCS:
https://marcs.helmholtz-munich.de/
文章链接:
https://www.nature.com/articles/s41586-024-07141-5


