







♦ 邻近标记技术在糖-蛋白质相互作用研究中的应用

Scripps Research Mia L. Huang 团队2023年在 Current Opinion in Chemical Biology 发表了review 文章。
这篇文献的主题是邻近标记技术在糖-蛋白质相互作用研究中的应用。糖-蛋白质相互作用是指一种生物分子之间的识别和结合过程,它在细胞信号传导、免疫调节、细胞粘附等生理功能中起着重要作用。但是,由于糖-蛋白质相互作用通常是暂时性的、弱力的、多价的,并且受到细胞环境和糖链结构多样性的影响,因此很难通过传统的生化方法来捕捉和分析。
邻近标记技术是一种新兴的实验方法,它可以通过在一个感兴趣的蛋白质(例如一个能够识别特定糖链结构的蛋白质)附近标记其他生物分子(例如与之结合的糖修饰蛋白质),来揭示其潜在的相互作用伙伴。邻近标记技术通常利用一些非人类酶(例如APEX或BioID)来在活细胞中催化生物素与邻近蛋白质之间的共价连接,然后通过亲和纯化和质谱分析来鉴定被标记的蛋白质。
https://doi.org/10.1016/j.cbpa.2022.102233
♦ 邻近标记技术

文章回顾了近距离标记技术的原理、优势和局限,并举例说明了它在不同领域和系统中的应用,如信号转导网络、酶-底物相互作用、动态过程和活体组织中的相互作用研究。文章还讨论了邻近标记技术面临的挑战和未来发展方向。
https://www.nature.com/articles/s41592-020-01010-5
♦ 免疫治疗模型

Memorial Sloan Kettering Cancer Center, New York 的 Timothy A. Chan 等人在2022年的 Nature Biotechnology 发表了题为Improved prediction of immune checkpoint blockade efficacy across multiple cancer types 的文章
这是一篇关于使用机器学习模型预测免疫检查点抑制剂(ICB)反应的研究。免疫检查点抑制剂是一类能够激活免疫系统对抗肿瘤的药物,但是只有部分患者能够从中受益,而目前的决策方法准确性有限。
文章介绍了一个基于随机森林算法的机器学习模型,该模型综合了来自一个大规模队列(MSK-IMPACT)的基因组、分子、人口统计和临床数据,用于预测1,479名接受ICB治疗的16种不同癌症类型患者的ICB反应。
文章展示了该模型在回顾性分析中表现出高灵敏度和特异度,并且能够预测不同癌症类型患者的总生存期和无进展生存期。文章还比较了该模型与基于肿瘤突变负荷(TMB)的预测方法,发现该模型明显优于TMB,而TMB是目前唯一被美国食品药品监督管理局(FDA)批准用于这一目的的指标。
♦ 空间表观多组学分析

耶鲁大学和卡罗琳斯卡医学院合作提出了一种新的技术,即空间表观遗传-转录组共分析技术,该技术整合了ATAC-seq、RNA-seq和CUT&Tag,可以通过检测出在某个时间点下,基因组上哪些区域是开放的并且易于被转录因子结合或修饰,分析细胞的表观遗传修饰和转录组,从而更好地理解细胞的功能和组织结构。
该技术采用了一种组织冷冻切片 、荧光原位杂交和高通量测序的组合方法,可以将组织样品的表观遗传修饰和转录组信息分析到亚细胞水平,并对组织的空间结构和异质性进行深入研究,目前该技术达到每个组织切片10000像素。
作者通过该技术对小鼠肝脏、脾脏和大脑进行了研究,鉴定了一些重要的表观遗传修饰和转录调控元件,并发现了这些组织中的细胞类型和功能的异质性。该技术的应用为深入理解细胞功能和组织结构,并为后续探索疾病发生机制提供了新的手段。
♦ 高灵敏度和深度的免疫肽组

人白细胞抗原(HLA)的鉴定分析常使用质谱来进行。为了达到足够的鉴定深度,常常需要使用离线分馏的方式、对相同的样本多次进样。这种方法往往需要比较多的样本起始量,而这对较为珍贵临床样本有时候不切实际。
接下来,他们优化了样本起始量,并实现了从低至1E6个A375细胞的起始量鉴定出约800条肽,其中包括了来自CTA、nuORF等生物学意义很强的HLA-I肽。该方法也体现出了timsTOF系列产品在高灵敏鉴定中的巨大能力。
♦ PPIs

文章介绍了蛋白质-蛋白质相互作用(PPIs)领域的最新进展。
蛋白质-蛋白质相互作用是指两个或多个蛋白质分子之间的结合和相互作用,它们在生物体中的许多过程中起着关键作用,例如抗体-抗原结合、细胞信号传导、基因表达和调控、病毒自组装等。蛋白质-蛋白质相互作用的研究涉及多个学科领域,如蛋白组学、生物物理学、生物化学、生物信息学和药理学,它们利用各自的方法和工具来探索蛋白质的结构、相互作用和功能之间的联系,并利用这些知识来设计新型药物。
这个专刊包含了8篇原创文章和4篇综述文章,涵盖了不同的蛋白质和不同的实验技术以及计算方法,展示了该领域的多样性和活跃性。具体来说:
Shoshani et al. 研究了人类Na+ , K+ -ATPase 的β2 亚基与哪些蛋白质结合,从而介导星形胶质细胞与神经元之间的粘附。他们发现该蛋白质在非星形胶质细胞上表达时可以与自身结合,表现出同源性细胞粘附分子的特征。
Eronina et al. 研究了一种小分子热休克蛋白αB-crystallin 与糖原磷酸化酶b 的相互作用。αB-crystallin 作为分子伴侣可以抑制糖原磷酸化酶b 的聚集。该相互作用改变了两种蛋白质的三级和四级结构。作者还发现甜菜碱可以增强抗聚集活性,而精氨酸则可以降低抗聚集活性。
Gorshkov et al. 分析了不同种类的Svx-like 蛋白,它们是植物致病菌属 Pectobacterium 的毒力因子。他们建立了 Pectobacterium atrosepticum 的 Svx 蛋白的原子模型,并讨论了其与α-糖基化蛋白之间可能的相互作用。
Ilinskaya et al. 比较了不同 Bacillus 物种中同源核糖核酸酶(RNases)与其抑制剂 barstar 之间的相互作用以及对不同 RNA 底物的催化活性。他们发现即使是微小的结构变化也会对两种蛋白质的稳定性和功能活性产生显着影响。
♦ 蛋白质相互作用网络

它还总结了从这些方法中识别和定量相互作用的计算挑战和解决方案,如处理携带污染、背景污染、假阳性相互作用、简并肽段、化学计量变化和光谱峰饱和等问题。它最后强调了这些技术和流程的当前局限性和未来发展方向。
♦ 用表观遗传标记鉴定甲状腺结节

甲状腺超声检查的结节患病率约为 70%,其中只有约 5% 为恶性。准确评估和诊断甲状腺结节的良恶性对医生采取适当的临床管理至关重要。超声成像结合细针穿刺 (FNA) 活检是诊断甲状腺结节的主要方法,但有约 20%-30% 的结节诊断不确定。
南方科技大学的邢明照教授和上海十院等8家医院合作,利用表观遗传学标记物鉴定甲状腺结节的良恶性,结果今年2月20日发表在 Journal of Clinical Oncology 上。
作者使用的技术叫做定量显色印迹基因原位杂交技术(QCIGISH),可以使用探针结合预先选定的印迹基因的pre-mRNA 非编码内含子区域,探针结合后在普通显微镜下能看到明显的红色或棕色点,然后通过手动和图像识别软件程序自动计数每个细胞内的表达信号,表达信号与恶性程度成正相关。
♦ 质谱结合平衡透析系统地发现变构

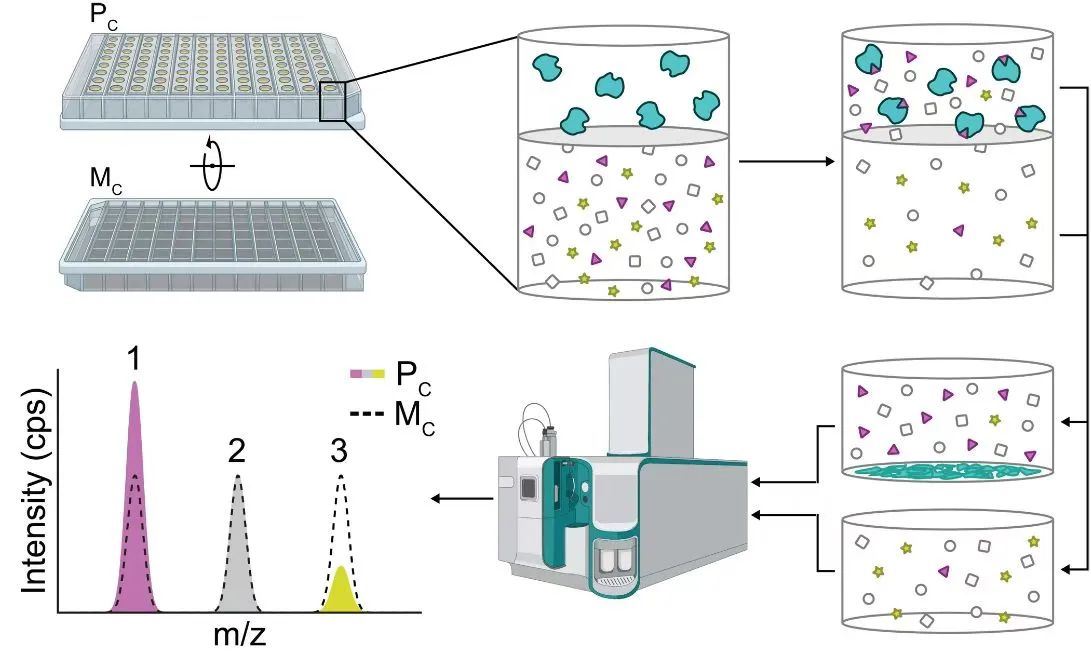
代谢物和蛋白的相互作用网络对细胞的生物学功能产生重要影响,这种相互作用通常是低亲和力(通常是uM到mM)。
美国犹他大学 Jared Rutter 团队于2023年3月9日在Science杂志上发表了一篇题为 Protein-metabolite interactomics of carbohydrate metabolism reveal regulation of lactate dehydrogenase 的文章,作者提出了一种称为 “质谱结合平衡透析系统地发现变构”(mass spectrometry integrated with equilibrium dialysis for the discovery of allostery systematically,MIDAS)的方法,用于分析这些低亲和力的蛋白质-代谢物相互作用(PMIs)。
该方法原理是平衡透析的生物物理原理。简单地说,装置分两层蛋白质室(Pc)装载纯化的蛋白和代谢物室(Mc)装载代谢物。半透膜允许代谢物自由通过,而蛋白不能通过。将纯化的蛋白质与代谢物进行结合,系统达到相对平衡。
通过沉淀去除蛋白质,对Pc和Mc中的代谢物进行采样,并使用流动注射分析-质谱(flow injection analysis–mass spectrometry,FIA-MS)对来自两个室的代谢物的相对丰度进行量化。Pc中相对于Mc(点峰)代谢物丰度的增加(1)或减少则被定义为PMI,两处丰度相同则定义为无PMI。
♦ GoDig

由于靶向定量准确性和敏感性会受到共洗脱蛋白数量的影响,因此探索了最佳靶向鉴定条件为2小时内243个蛋白。他们利用该方法可以在60小时内鉴定480种多样性远交高脂喂养小鼠的肝脏样本的一组靶向蛋白(激酶、脂代谢、脂滴形成相关蛋白),并与已有小鼠肝脏样本的代谢组数据库进行比对,展示基因变异对蛋白表达和脂质调节的影响。
♦ 宏蛋白质组方法分析溃疡性肠炎患者的粪便微生物组

本文2022年3月发表在MCP,采用宏蛋白质组方法分析了8个健康人与10个溃疡性肠炎(UC)患者的粪便微生物组,同时进行了16S rRNA 基因测序,比较了两种技术方法所鉴定的微生物群落结构的较大差异,部分原因是宏蛋白质组可检测到宿主、真菌、病毒和食物的肽段。
宏蛋白质组检测到健康人与UC患者间显著差异的176个protein groups,并通过GO分析揭示了UC相关的几种蛋白质功能,其中发现UC患者中丝氨酸型肽链内切酶活性高,其功能最为突出。
♦ 血浆样本预测非小细胞肺癌患者在经历免疫检查点抑制剂治疗后反应的潜在标志物

免疫检查点抑制剂(immune checkpoint inhibitors)例如程序性细胞死亡蛋白-1(PD-1)和程序性死亡配体- 1(PD-L1)作为非小细胞肺癌的治疗方案。作为体外活检,液体活检分析外周血中游离的DNA,蛋白质含量水平等,用于潜在的生物标志物。
2022年6月,以色列 Yuval Shaked 团队在 journal of immunotherapy of cancer 发表了一篇研究论文(题目如上),使用血浆样本预测非小细胞肺癌患者在经历免疫检查点抑制剂治疗后反应的潜在标志物。来自于1号和2号队列的143位经历单一疗法或与联合疗法的IIIB-IV期非小细胞肺癌患者,作为研究对象。
内容主要包括5个部分:
临床信息和治疗反应之间的相关性分析。非小型细胞癌患者在经历免疫检查点抑制剂治疗后,通过3个月实体瘤的疗效为标准,将病人分成响应型患者(病情稳定,实体瘤部分或全部缓解)和非响应型患者(病情持续发展)。作者比较了以下几种临床数据和两种反应型患者的关系,年龄;PD-L1的肿瘤比例评分;患者治疗效果;KRAS和TP53突变情况等。
基于血浆样本对治疗反应的预测。作者将来自两个队列的数据分成训练集(n=72),验证集(n=36)和测试集(n=35),基于XGBoost算法结合蛋白质组学信息,临床信息进行分析,以开始治疗后的2-6周的时间点,判断患者属于治疗响应型或治疗非响应型。模型在测试集的AUC数值为0.79。作者在这个基础上又研究了不同临床因素对预测模型的潜在影响。
治疗后非小细胞肺癌患者的血浆蛋白质表达图谱。作者选择包含更多临床信息的2号队列作为研究对象,基于蛋白质组学的数据进行一致性聚类分析产生不同的3种分组。研究了不同分组下蛋白表达情况和信号通路。
响应型患者和非响应型患者之间的差异表达蛋白。并结合前人文献报道查看差异表达中的蛋白在人类蛋白图谱数据库的表达情况和功能分析。
♦ 药效和毒性预测

最后作者纳入临床ORR、预测的IC50、预测的毒性这3个特征,通过10折交叉验证建立机器学习模型预测药物获批可能性,发现基于ORR的预测模型AUC可以达到0.83,但不同折之间AUC分布范围大;而模型中同时纳入ORR、预测的IC50、预测的毒性后,AUC提升到0.89且不同折之间AUC更趋近;提示对IC50和毒性预测的意义。


