风乍起,吹皱一池春。
平静的小池塘迎来了风雨,水面渐起涟漪。
想象一下,生物体的细胞就像一个宁静的池塘,而各种 “微扰” 则如同大小不一、时间各异的雨滴落入其中。这些 “雨滴” 在生命之池中激起涟漪,形成复杂的波纹交织:有的相互叠加,级联放大效应;有的相互抵消,呈现出丰富的动态变化。
在微观层面,这些涟漪效应直接体现为蛋白质的多维度变化:表达丰度的起伏、亚细胞定位的迁移、蛋白质间相互作用的重组、以及翻译后修饰的调节。这些微观变化如同蝴蝶效应,最终在宏观上引发多样化的表型变化。
11月1日,西湖大学医学院 / 生命科学学院 / 西湖实验室 / 未来产业研究中心郭天南团队,联合哈佛医学院 Chris Sander、苏黎世联邦理工学院(ETH Zürich)Peter Bühlmann、Ruedi Aebersold ,在 Cell Genomics 发表了一篇 Perspective 文章。
西湖大学生命科学学院博士后钱鎏佳、西湖大学医学院助理研究员孙瑞为文章共同作者。

- 提纲挈领 -
文章围绕AI赋能的微扰蛋白质组学(AI-empowered perturbation proteomics)在复杂生物系统研究中的前景,提出了一种全新的PMMP研究流程(微扰-测量-建模-预测),以应对当前系统生物学领域面临的数据不足和建模局限。
通过引入多种生物、化学和物理层面的微扰手段,并结合高通量蛋白质组学、转录组学和代谢组学等多模态数据,研究者能够更加精准地预测生物系统对微扰的响应。利用AI模型,微扰蛋白质组学可以鉴定药物机制、探索未充分研究蛋白质的功能、预测扰动响应,比如单药敏感性及双药协同作用等,进而协助设计个性化治疗方案。
文章也指出,当前的挑战在于数据获取的成本及整合不同数据类型的复杂性,未来需通过多学科合作与数据标准化来实现临床应用的可行性。
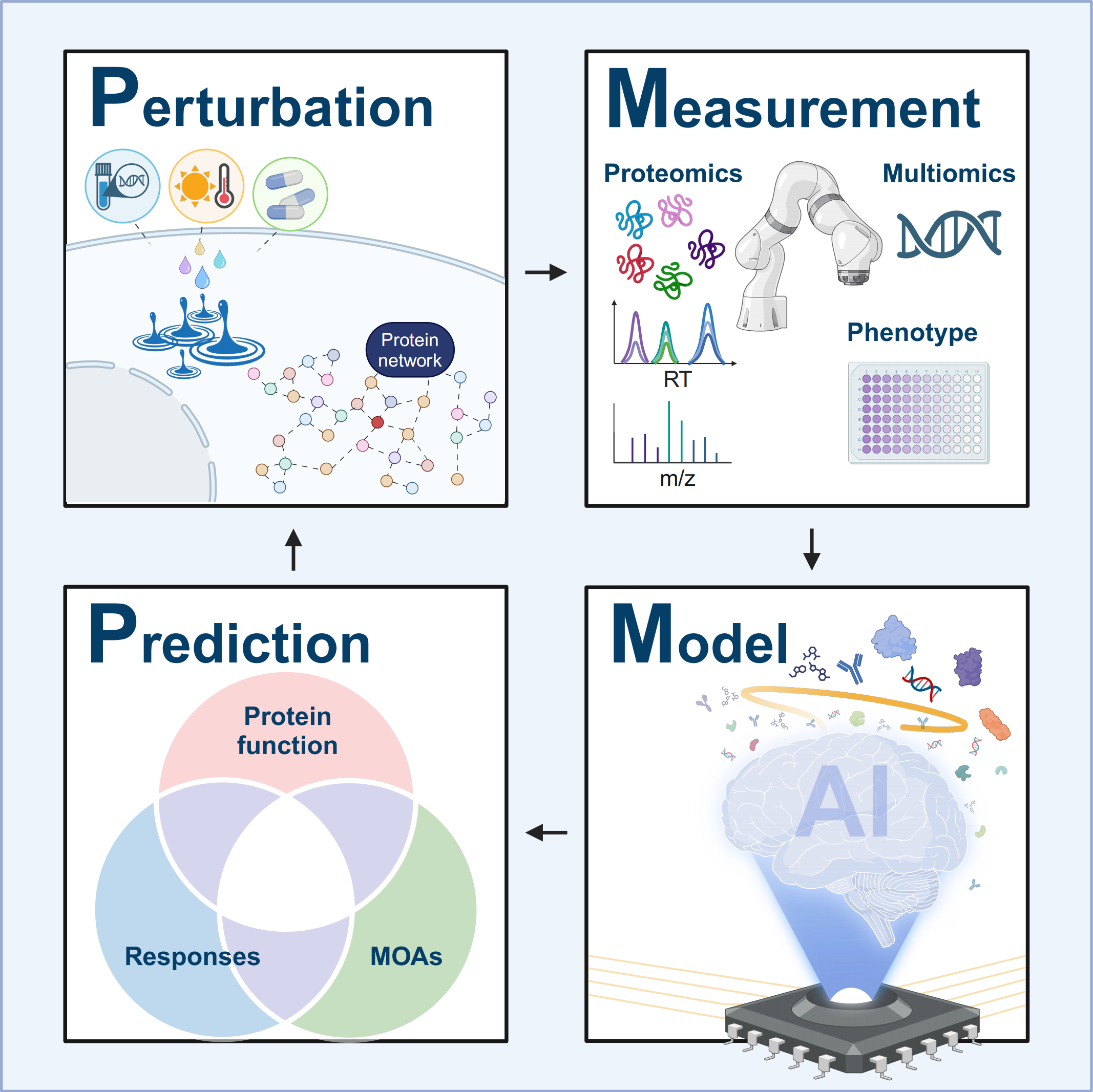
图文摘要
传统的系统生物学面临的一个主要障碍是缺乏蛋白质水平的全面微扰数据。微扰蛋白质组学(perturbation proteomics)旨在通过生物、化学和物理等不同的 “扰动” 手段,结合蛋白质的表达、修饰、交互、定位和表型变化,来揭示生物系统的响应模式和机制。
PMMP流程(Perturbation, Measurement, Modeling to Prediction,即扰动-测量-建模-预测)是一个系统化的框架。该流程以扰动实验为起点,通过对系统性数据的多层次测量,结合机器学习和深度学习等AI模型,最终实现对生物系统响应的精确预测。
具体而言,研究者利用不同种类的干扰(生物、化学或物理)来触发蛋白质组的变化,再通过大规模蛋白质组学数据的测量,构建模型来预测系统的响应。这一流程不仅能够揭示未知蛋白质的功能,还能更好地理解药物机制和个性化治疗的可能性。
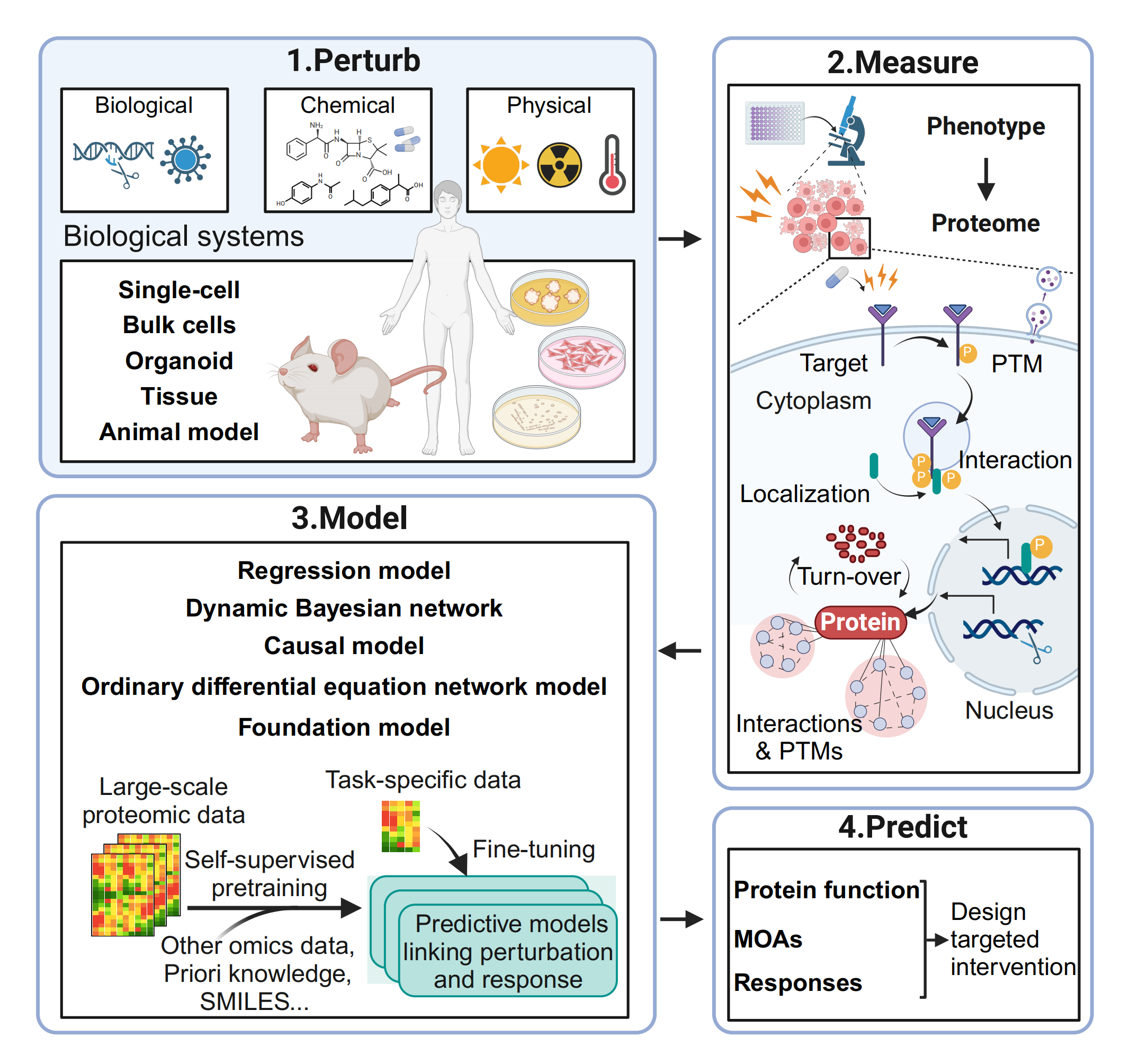
用于微扰蛋白质组学的PMMP流程(扰动-测量-建模-预测)
通过基因编辑、微生物感染、药物处理以及物理干预(如光照、温度、辐射)等多种方法对不同的生物系统(包括细胞、类器官、组织和动物模型)进行扰动。随后分析这些系统在表型特征和蛋白质层面特性的动态变化。蛋白质水平的测量可涵盖广泛的内容,包括对靶蛋白的直接影响、蛋白质-蛋白质相互作用和蛋白定位的改变、翻译后修饰和蛋白质周转的变化以及下游蛋白网络中的广泛修饰。接着,基于这些测量数据构建机器学习模型,旨在预测蛋白质功能、作用机制和系统对干扰的响应。这种预测能力对于制定精确的治疗干预措施以及规划系统生物学研究中的协同实验方法等至关重要。
构建PMMP流程面临的一个重要挑战在于如何处理复杂生物系统的动态性和多样性。有趣的是,研究人员们从物理学中得到了启发。
在理论物理学中,有一个经典笑话 Spherical cow(真空中的球形奶牛)。它源自这样一种假设:在研究复杂系统时,研究人员可能会简化条件到不切实际的程度(比如把形状复杂的奶牛假设成一个理想的球体,并且置于真空中),以便更容易地进行数学计算和建模。

Spherical cow, 图源:wikipedia
Spherical cow 代表了对复杂现象的过度简化模型,而这种过度简化可能导致对系统实际特征的忽视。在PMMP流程中,扰动实验的设计和引入是为了打破 Spherical cow 的局限性,通过不断迭代、添加新的扰动信息(即不断往 “简化模型” 中纳入更多的细节),使得模型的表现更贴近真实的生物系统。
在这一过程中,扰动(perturbations)作为 “催化剂” 来测试真实系统的复杂反应,可揭示出简化模型可能忽略的信息。
物理学中的 “扰动理论”(Perturbation theory)提供了模型改进的框架,而这种方法在系统生物学中也同样适用——通过迭代增加扰动信息,模型可以逐步精细化,从而提高对复杂系统的理解和预测能力。
文章指出,成功的微扰蛋白质组学分析流程不仅依赖高质量的蛋白质组学数据,还需要结合其他相关但独立的数据集,如转录组学、代谢组学、染色质状态(如DNAseI超敏筛查和ATACseq)、化合物的SMILES结构、蛋白质相互作用和调控网络,以及成像数据等。多模态数据的融合有助于消除生物系统中的噪声和数据缺失问题,同时为机器学习模型提供更丰富的信息背景。这种数据整合能够揭示新的生物学模式和因果关系,有助于发现生物标志物和药物靶点。
扰动实验由于可以控制变量,因此更适合进行因果关系的推断,而观察性研究(observational studies)在无法进行实验扰动时则提供重要的自然状态数据。尽管观察性研究因未观察到的混杂因素而存在局限性,但其对理解人类疾病和基因-环境交互效应等仍有重要意义。研究人员在文中指出,将扰动和观察数据结合,能够在更广泛的生物背景中验证和生成假设,从而为复杂生物系统的研究提供更全面的理解。
AI技术(尤其是深度学习)的引入将极大地推动微扰蛋白质组学的发展。AlphaFold2等AI模型在蛋白质结构预测方面取得了突破性进展,但真实生物系统中蛋白质功能的复杂性远超静态结构预测。
为应对这一挑战,基础模型等大型预训练模型展现出巨大潜力。例如,Geneformer模型通过3000万个单细胞转录组数据预训练,成功预测了心肌病的潜在靶点。然而,基础模型并非万能,我们仍需开发专门针对分子数据复杂性的模型。
另外,因果表示学习是一个极具前景的方向,有望实现对未见蛋白组扰动及其表型影响的预测。但目前公共数据库中缺乏足够的微扰蛋白质组学数据来构建大型预训练模型。对此,研究人员表示,新一代蛋白质组学技术(Next-generation proteomics technologies),如单细胞蛋白质组分析及多重标记技术,为大规模数据的生成提供了支持,从而有望解决数据稀缺的问题。
在文章最后,研究人员展望了微扰蛋白质组学在个性化治疗、药物研发和未充分研究蛋白质的功能研究中的应用潜力。同时,研究人员强调,未来需要跨学科合作,建立标准化的数据共享平台和分析框架,以推动微扰蛋白质组学的临床转化应用。
总结
文章表明,微扰蛋白质组学结合AI和多模态数据,为理解和预测生物系统在不同扰动下的响应提供了前所未有的可能性。通过系统化地测量和建模扰动响应,研究者可以更全面地探索蛋白质功能、药物作用机制以及疾病的分子基础。这种方法的突破将为个性化医学和生物工程带来新的研究路径,但在此过程中,数据获取的规模化和集成化仍然是一个重要挑战。