♦ 一种用于无需结构修饰鉴定小分子化合物靶点的方法

图源:文章截图
作者开发了一种用于无需结构修饰鉴定小分子化合物靶点的方法,即将两年前Mingliang Ye组的溶剂诱导蛋白质沉淀(SIP)方法与TMT标记定量蛋白质组学相结合,提出了溶剂诱导蛋白质组学分析(SIPP)方法来鉴定小分子与蛋白质的相互作用,可以潜在地揭示小分子化合物的药理机制。
SIPP方法通过改变有机溶剂的浓度诱导细胞蛋白质的变性和沉淀,从而得到目标蛋白质的变性曲线, 并确定蛋白质的特定熔点浓度(Cm)作为预测候选靶点的关键指标。SIPP方法展现了深入的蛋白质组信息和比SIP方法更高的小分子活性化合物靶点鉴定效能, 不仅成功地鉴定了staurosporine(激酶全抑制剂)和methotrexate(治疗多类型癌症)的已知靶点,还预测了staurosporine的非激酶蛋白靶点。
作者比较了SIPP方法,SIP方法与TPP(热蛋白质组学)方法所鉴定到的Hela细胞中靶标蛋白质, 数据在Q Exactive Plus上采集, MaxQuant(version 2.0.3.0)分析,SIPP方法鉴定数目约为SIP的2.5倍(55/20),虽然略少于TPP方法(55/60),但有35种不同于TPP, 表明SIPP方法可以作为现有无需修饰的蛋白质组学方法鉴定活性化合物靶点技术的补充, 若采用更高灵敏度的质谱分析也许可以得到更多的target proteins。
https://analyticalsciencejournals.onlinelibrary.wiley.com/doi/10.1002/pmic.202200281
♦ 尿液蛋白与肿瘤生长、恶性程度和转移相关

图源:论文截图
赵晓航/孙伟团队采用TMT技术分析了健康对照、无转移结直肠癌、淋巴结转移结直肠癌以及远端转移结直肠癌患者的尿液样本,发现尿液蛋白与肿瘤生长、恶性程度和转移相关。
接下来在82例独立队列中进行了PRM验证,并通过随机森林的方法构建了3个尿液蛋白组成的结直肠癌诊断模型以及由4个尿液蛋白组成的转移风险预测模型,发现尿液诊断标志物组合与粪便免疫化学检测结果联用能显著提升结直肠癌诊断的敏感性(从60%提高到85%),同时转移风险预测标志物组合可以将血清癌胚抗原的检测灵敏度从58.1%提高到82.9%。
随后在3个独立队列样本中进行免疫组化验证,证实尿液中3个诊断相关蛋白的异常改变与组织染色结果一致。
https://pubmed.ncbi.nlm.nih.gov/35589723/
♦ AI辅助筛选抗菌肽

图源:论文截图
中科院微生物所王军老师、陈义华老师团队文章。
在具有生物活性的次级代谢产物中,短肽因其高度多样性和广泛的生物活性谱而受到广泛关注,尤其是来自细菌的大量抗菌肽已被用于治疗细菌、真菌和病毒感染,甚至癌症。从理论上讲,大量来自人类肠道微生物群的潜在抗菌肽(Antimicrobial Peptides, AMPs)可以作为抗感染的候选来源。人类肠道微生物组编码多种AMPs,但AMPs的长度较短,这对计算预测提出了挑战。
该文结合了多种自然语言处理神经网络模型,包括LSTM, Attention和BERT,以形成从人类肠道微生物组数据中识别候选AMPs的统一pipeline。在被鉴定为候选AMPs的2349个序列中,216个是化学合成的,其中181个显示出抗菌活性(阳性率> 83%)。
这些肽中的大多数与训练集中的AMPs具有小于40%的序列同源性。对鉴定到的11种最有效的AMPs的进一步实验验证,显示了它们对抗生素抗性、革兰氏阴性病原体的高效性,并证明了在针对细菌性肺部感染的小鼠模型中这些AMPs能够将细菌载量降低10倍以上。
该研究展示了机器学习方法在从宏基因组数据中挖掘功能肽和加速发现有前途的AMPs候选分子以进行深入研究方面的潜力。
♦ 基于序列衍生的物理化学性质和独立于生物丰度的可检测性预测
图源:论文截图
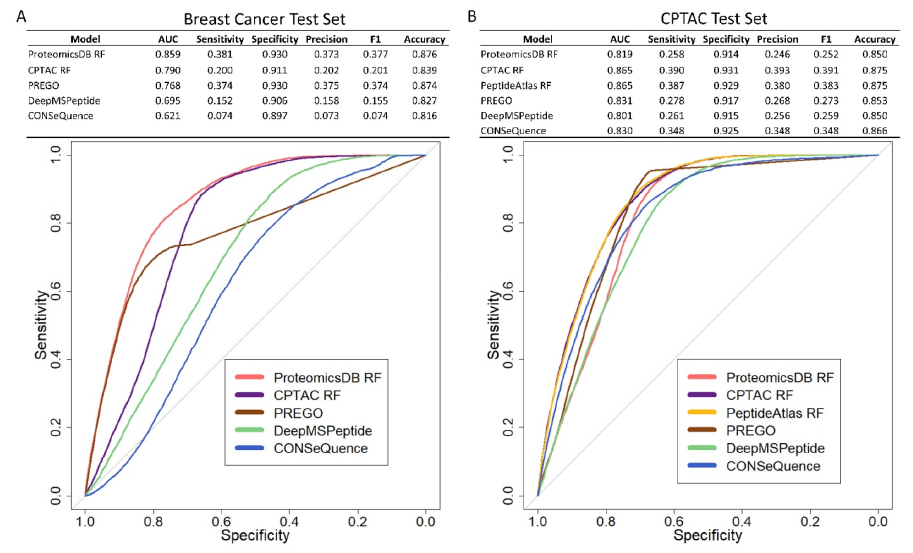
图源:文章Figure
这篇文章提供了一个基于序列衍生的物理化学性质和独立于生物丰度的可检测性预测,返回所提供列表中每个蛋白质的top peptides。
该分类器利用ProteomicsDB(包含1479个(27/Nov/2020)人体实验,涵盖多种实验配置、量化方法和质谱类型)使用随机森林(RF)进行训练和特征选择。该方法主要可以用来优化质谱分析中的合成肽的选择,能够帮助更有效的选择合成肽,以提高质谱分析的准确性和灵敏度。该
R包还支持利用公开MS数据集进行处理,并管理数据集,可以更好的匹配特定的实验条件。这篇文章还与之前的几种方法:
PREGO Predictions:一种使用独立数据采集(DIA)来建立高响应肽段模型的方法,以用于定向蛋白质组学实验(https://www.sciencedirect.com/science/article/pii/S1535947620326426)
进行比较,使用两组数据集(breast cancer (BrCa) cohort;CPTAC databases),结果表明,与之前的模型相比,该R包提供的方法在各种数据集上预测可检测肽的精度有所提高(如上图文章Figure),并且根据ProteomicsDB中的观察结果提高了蛋白质top peptides的排名。
♦ 宏蛋白组蛋白推断

图源:文章截图
一篇宏蛋白组蛋白推断(protein inference)的文章。
由于大量同源蛋白的存在,宏蛋白质组领域的蛋白推断的挑战更大,这篇文章将来自于16S/metagenomics数据的物种丰度信息与肽段和蛋白的可信度信息结合,利用整合线性规划模型(integrative linear programming method)进行宏蛋白样本的protein inference。
它的核心思想是将protein inference转化为一个优化问题,即找到一个smallest subset of proteins,可以最好地 “解释” peptides和taxonomic abundances。
通过在不同dataset的benchmark test发现相比于其他的protein inference method (ProteinLP, PeptideProphet, DeepPep, PIPQ, and Sipros Ensemble),可以提高2%-30%的protein identification,同时accuracy在0.998。
另外这篇文章提到了包括基于统计学模型和machine/deep learning的多种protein inference的算法。
♦ 多模态质谱数据整合分析平台MZmine3

图源:论文截图
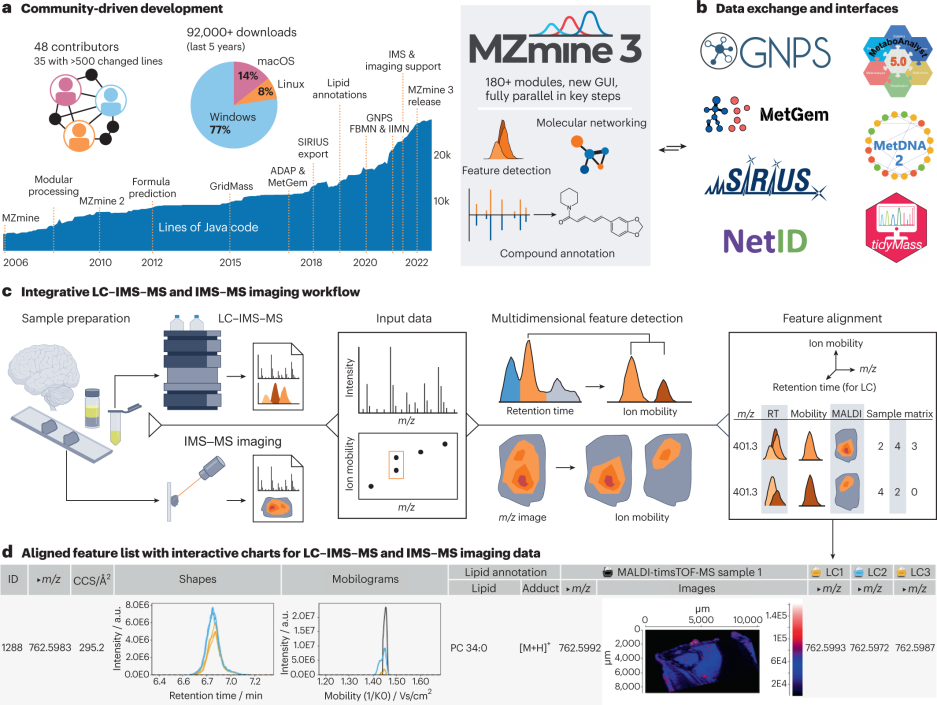
图源:文章Figure
3月1日Nature biotech发布了一篇correspondance,是一个多模态质谱数据整合分析平台MZmine3 ,可分析液相或气相色谱单质谱(LC-MS or GC-MS),ion mobility质谱(IMS)和质谱图像的数据,特别可以应用在联合IMS-MS图像和液相IMS-MS的方法为空间代谢组进行分析。
基于JAVA语言的社区开源项目MZmine初代在2004发布,十几年来不断增加新的工具如化合注释SIRIUS,统计分析工具MetaboAnalyst,也可以结合GNPS网络分析平台进行分析。基于PASEF的IMS-MALDI质谱( timsTOF fleX)的进展提高了MALDI成像质谱的灵敏度、分辨率和数据采集速度的临床大队列的应用成为可能,然而数据量和复杂性也随之增加,因而需要更强大的数据分析工具来分析新型的质谱数据。
MZmine3中对图像特征的 RT、m/z 和离子淌度值进行了特征对齐,并进一步整合进入空间组学数据,解决了大多数成像研究中缺少 MS2 数据的问题。MZmine3也对大数据队列进行了数据压力测试,仅需要 47 分钟便可分析8,273 个粪便样本。最新版本的 MZmine3可以从 https://www.mzmine.org下载。此项工作还有西湖大学工学院学生Miaoshan Lu参与。
♦ 一种用于无需结构修饰鉴定小分子化合物靶点的方法
图源:文章截图
作者开发了一种用于无需结构修饰鉴定小分子化合物靶点的方法,即将两年前Mingliang Ye组的溶剂诱导蛋白质沉淀(SIP)方法与TMT标记定量蛋白质组学相结合,提出了溶剂诱导蛋白质组学分析(SIPP)方法来鉴定小分子与蛋白质的相互作用,可以潜在地揭示小分子化合物的药理机制。
SIPP方法通过改变有机溶剂的浓度诱导细胞蛋白质的变性和沉淀,从而得到目标蛋白质的变性曲线, 并确定蛋白质的特定熔点浓度(Cm)作为预测候选靶点的关键指标。SIPP方法展现了深入的蛋白质组信息和比SIP方法更高的小分子活性化合物靶点鉴定效能, 不仅成功地鉴定了staurosporine(激酶全抑制剂)和methotrexate(治疗多类型癌症)的已知靶点,还预测了staurosporine的非激酶蛋白靶点。
作者比较了SIPP方法,SIP方法与TPP(热蛋白质组学)方法所鉴定到的Hela细胞中靶标蛋白质, 数据在Q Exactive Plus上采集, MaxQuant(version 2.0.3.0)分析,SIPP方法鉴定数目约为SIP的2.5倍(55/20),虽然略少于TPP方法(55/60),但有35种不同于TPP, 表明SIPP方法可以作为现有无需修饰的蛋白质组学方法鉴定活性化合物靶点技术的补充, 若采用更高灵敏度的质谱分析也许可以得到更多的target proteins。
https://analyticalsciencejournals.onlinelibrary.wiley.com/doi/10.1002/pmic.202200281
♦ 尿液蛋白与肿瘤生长、恶性程度和转移相关
图源:论文截图
赵晓航/孙伟团队采用TMT技术分析了健康对照、无转移结直肠癌、淋巴结转移结直肠癌以及远端转移结直肠癌患者的尿液样本,发现尿液蛋白与肿瘤生长、恶性程度和转移相关。
接下来在82例独立队列中进行了PRM验证,并通过随机森林的方法构建了3个尿液蛋白组成的结直肠癌诊断模型以及由4个尿液蛋白组成的转移风险预测模型,发现尿液诊断标志物组合与粪便免疫化学检测结果联用能显著提升结直肠癌诊断的敏感性(从60%提高到85%),同时转移风险预测标志物组合可以将血清癌胚抗原的检测灵敏度从58.1%提高到82.9%。
随后在3个独立队列样本中进行免疫组化验证,证实尿液中3个诊断相关蛋白的异常改变与组织染色结果一致。
https://pubmed.ncbi.nlm.nih.gov/35589723/
♦ AI辅助筛选抗菌肽
图源:论文截图
中科院微生物所王军老师、陈义华老师团队文章。
在具有生物活性的次级代谢产物中,短肽因其高度多样性和广泛的生物活性谱而受到广泛关注,尤其是来自细菌的大量抗菌肽已被用于治疗细菌、真菌和病毒感染,甚至癌症。从理论上讲,大量来自人类肠道微生物群的潜在抗菌肽(Antimicrobial Peptides, AMPs)可以作为抗感染的候选来源。人类肠道微生物组编码多种AMPs,但AMPs的长度较短,这对计算预测提出了挑战。
该文结合了多种自然语言处理神经网络模型,包括LSTM, Attention和BERT,以形成从人类肠道微生物组数据中识别候选AMPs的统一pipeline。在被鉴定为候选AMPs的2349个序列中,216个是化学合成的,其中181个显示出抗菌活性(阳性率> 83%)。
这些肽中的大多数与训练集中的AMPs具有小于40%的序列同源性。对鉴定到的11种最有效的AMPs的进一步实验验证,显示了它们对抗生素抗性、革兰氏阴性病原体的高效性,并证明了在针对细菌性肺部感染的小鼠模型中这些AMPs能够将细菌载量降低10倍以上。
该研究展示了机器学习方法在从宏基因组数据中挖掘功能肽和加速发现有前途的AMPs候选分子以进行深入研究方面的潜力。
♦ 基于序列衍生的物理化学性质和独立于生物丰度的可检测性预测
图源:论文截图
图源:文章Figure
这篇文章提供了一个基于序列衍生的物理化学性质和独立于生物丰度的可检测性预测,返回所提供列表中每个蛋白质的top peptides。
该分类器利用ProteomicsDB(包含1479个(27/Nov/2020)人体实验,涵盖多种实验配置、量化方法和质谱类型)使用随机森林(RF)进行训练和特征选择。该方法主要可以用来优化质谱分析中的合成肽的选择,能够帮助更有效的选择合成肽,以提高质谱分析的准确性和灵敏度。该
R包还支持利用公开MS数据集进行处理,并管理数据集,可以更好的匹配特定的实验条件。这篇文章还与之前的几种方法:
PREGO Predictions:一种使用独立数据采集(DIA)来建立高响应肽段模型的方法,以用于定向蛋白质组学实验(https://www.sciencedirect.com/science/article/pii/S1535947620326426)
进行比较,使用两组数据集(breast cancer (BrCa) cohort;CPTAC databases),结果表明,与之前的模型相比,该R包提供的方法在各种数据集上预测可检测肽的精度有所提高(如上图文章Figure),并且根据ProteomicsDB中的观察结果提高了蛋白质top peptides的排名。
♦ 宏蛋白组蛋白推断
图源:文章截图
一篇宏蛋白组蛋白推断(protein inference)的文章。
由于大量同源蛋白的存在,宏蛋白质组领域的蛋白推断的挑战更大,这篇文章将来自于16S/metagenomics数据的物种丰度信息与肽段和蛋白的可信度信息结合,利用整合线性规划模型(integrative linear programming method)进行宏蛋白样本的protein inference。
它的核心思想是将protein inference转化为一个优化问题,即找到一个smallest subset of proteins,可以最好地 “解释” peptides和taxonomic abundances。
通过在不同dataset的benchmark test发现相比于其他的protein inference method (ProteinLP, PeptideProphet, DeepPep, PIPQ, and Sipros Ensemble),可以提高2%-30%的protein identification,同时accuracy在0.998。
另外这篇文章提到了包括基于统计学模型和machine/deep learning的多种protein inference的算法。
♦ 多模态质谱数据整合分析平台MZmine3
图源:论文截图
图源:文章Figure
3月1日Nature biotech发布了一篇correspondance,是一个多模态质谱数据整合分析平台MZmine3 ,可分析液相或气相色谱单质谱(LC-MS or GC-MS),ion mobility质谱(IMS)和质谱图像的数据,特别可以应用在联合IMS-MS图像和液相IMS-MS的方法为空间代谢组进行分析。
基于JAVA语言的社区开源项目MZmine初代在2004发布,十几年来不断增加新的工具如化合注释SIRIUS,统计分析工具MetaboAnalyst,也可以结合GNPS网络分析平台进行分析。基于PASEF的IMS-MALDI质谱( timsTOF fleX)的进展提高了MALDI成像质谱的灵敏度、分辨率和数据采集速度的临床大队列的应用成为可能,然而数据量和复杂性也随之增加,因而需要更强大的数据分析工具来分析新型的质谱数据。
MZmine3中对图像特征的 RT、m/z 和离子淌度值进行了特征对齐,并进一步整合进入空间组学数据,解决了大多数成像研究中缺少 MS2 数据的问题。MZmine3也对大数据队列进行了数据压力测试,仅需要 47 分钟便可分析8,273 个粪便样本。最新版本的 MZmine3可以从 https://www.mzmine.org下载。此项工作还有西湖大学工学院学生Miaoshan Lu参与。