








文献目录
1 SUM-PAINT:单蛋白分辨率的神经元空间蛋白质组学
2 scPROTEIN:用于单细胞蛋白质组嵌入的多功能深度图对比学习框架
3 DIP-MS:用于解构蛋白复合物的超深度相互作用蛋白质组学
4 Review: 基于组学的脂肪肝疾病生物标记物的发现机遇与障碍
5 内源性相分离蛋白质鉴定新策略
6 肾脏多组学的基因推断揭示新的血压和高血压靶点
7 通过深度学习预测糖肽片段质谱
8 One-Tip:全面覆盖最小细胞和单个胚胎的蛋白组学新方法
9 ConDiGA:在宏蛋白组学中准确构建蛋白质序列数据库
10 TBK1 p.E696K突变导致小鼠渐冻症
一起来看看本周蛋白组学精选优质文献吧!
1.(Cell,IF: 64.5)SUM-PAINT:单蛋白分辨率的神经元空间蛋白质组学
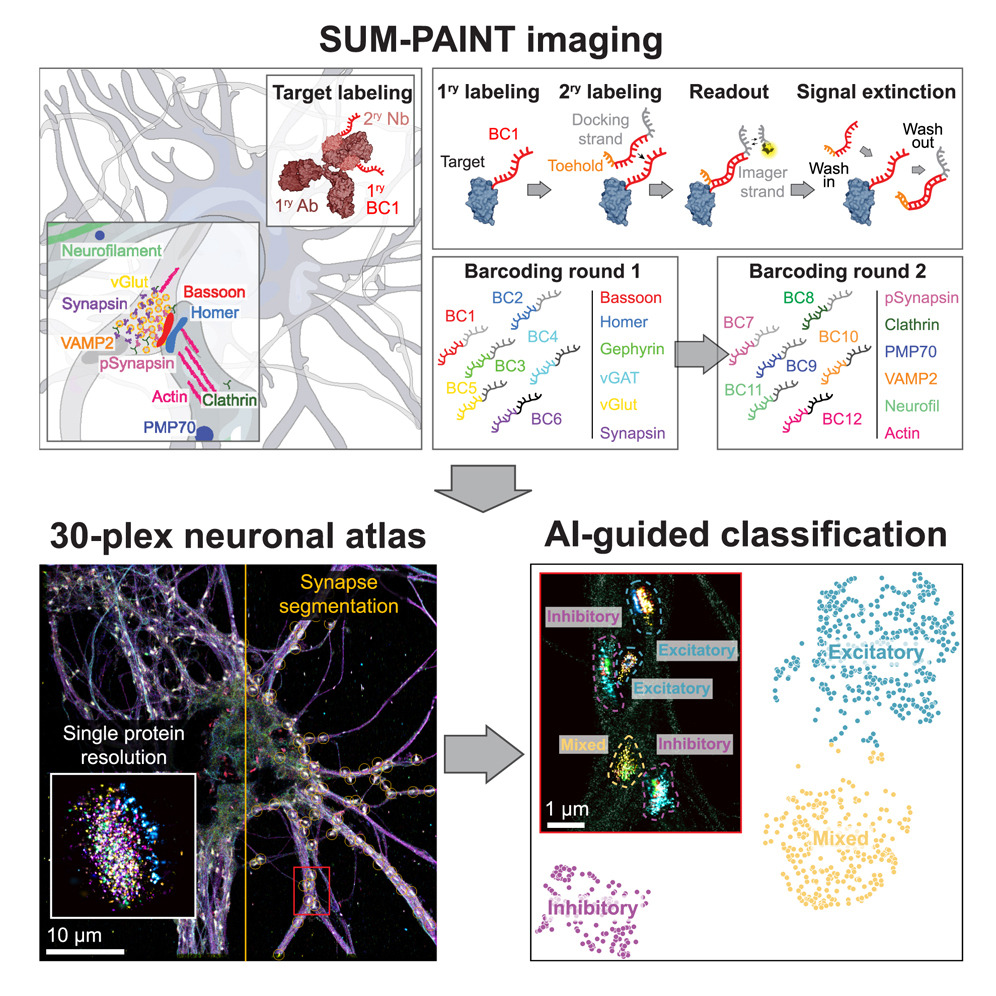
传统的蛋白质组学方法虽然在量化核酸和蛋白质丰度方面取得了显著进展,但要全面理解生物过程和功能,就需要空间组学技术,来提供绝对定量和亚细胞定位。
3月28日,马克斯·普朗克生物化学研究所的Ralf Jungmann团队及哥廷根大学医疗中心的Eugenio F. Fornasiero团队合作在Cell上发表了最新研究:Spatial proteomics in neurons at single-protein resolution。
文章介绍了一种名为SUM-PAINT的新型成像技术,利用二级标记的DNA-PAINT实现了高度多重、单蛋白水平的超分辨率成像,为构建单蛋白水平的细胞图谱提供了新途径。该研究也存在一定的局限性,例如SUM-PAINT受到亲和试剂的限制,还需要光学切片来实现对细胞的全面成像。
https://www.cell.com/cell/fulltext/S0092-8674(24)00248-4#%20
2.(Nat Methods,IF:48)scPROTEIN:用于单细胞蛋白质组嵌入的多功能深度图对比学习框架

3月19日,腾讯AI Lab和南开大学人工智能学院的研究人员在 Nature Methods合作发表了新的研究 scPROTEIN: a versatile deep graph contrastive learning framework for single-cell proteomics embedding,旨在解决单细胞蛋白质组学数据分析中的一系列挑战。
文章介绍了一种名为scPROTEIN的单细胞蛋白质组学数据分析框架,能够有效处理数据不确定性、批次效应和数据缺失问题,并在细胞聚类、数据整合、标签转移和空间分析等方面展现了优越性能。
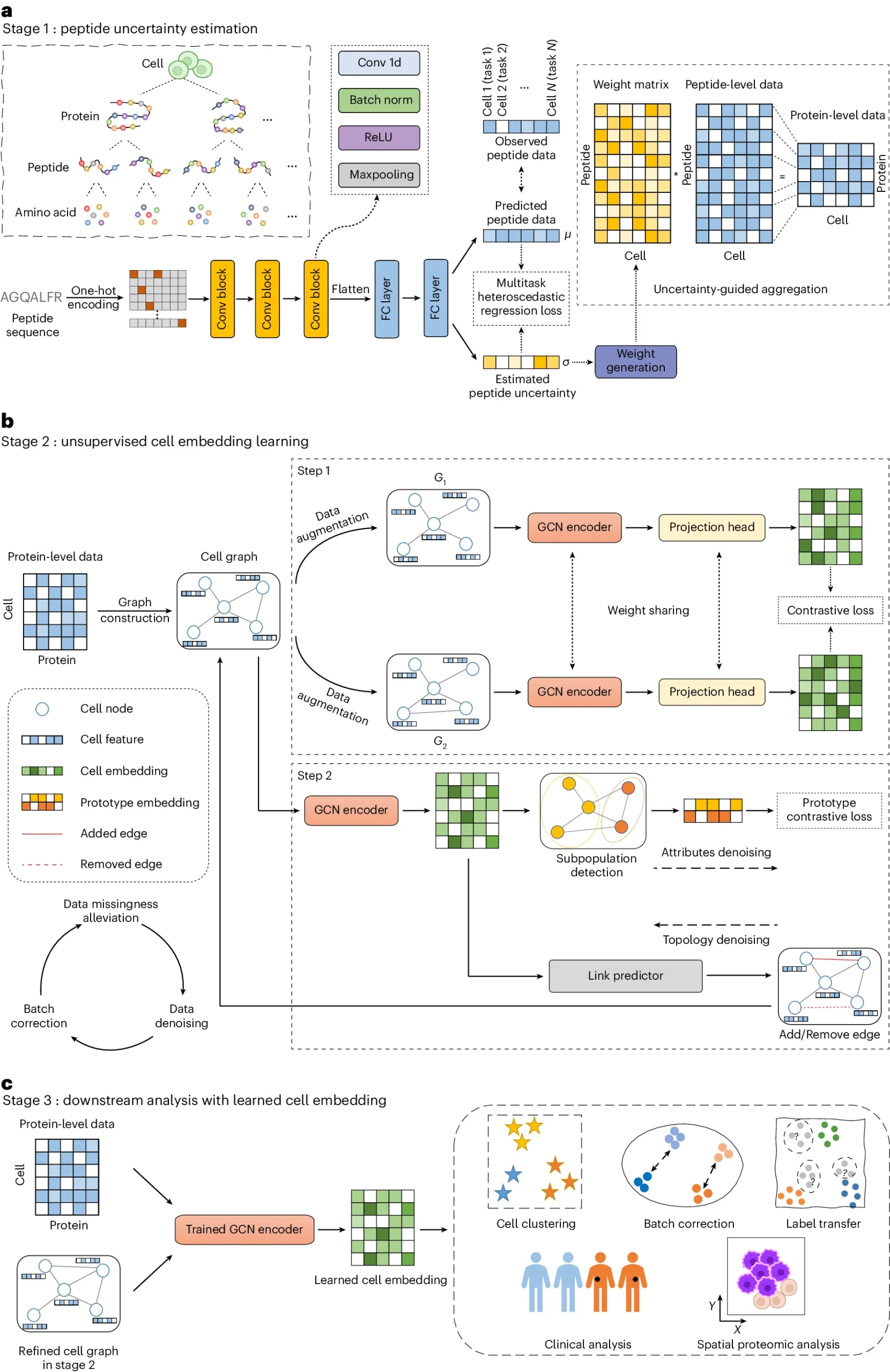
scPROTEIN的整体框架包括三个阶段的工作流程。
第一阶段通过多任务异方差回归模型估计肽段信号的不确定性,将其聚合到蛋白质水平。第二阶段构建细胞图,利用图对单细胞蛋白质数据进行学习和降噪。第三阶段利用经过训练的图卷积网络编码器学习细胞嵌入,用于各种下游任务。
这一框架能够有效地处理肽段信号的不确定性、数据缺失、批次效应和数据噪声等问题。该方法在多种单细胞蛋白质组数据集上得到了验证,并展现了广泛的应用前景。
https://www.nature.com/articles/s41592-024-02214-9?utm_medium=external_display&utm_source=stork&utm_content=email&utm_term=null&utm_campaign=CONR_JRNLS_AWA1_CN_CNPL_0034V_STKRE
3.(Nat Methods,IF:48)DIP-MS:用于解构蛋白复合物的超深度相互作用蛋白质组学

近期,苏黎世联邦理工学院分子系统生物学研究所的 Matthias Gstaiger 团队引入了一种称为 “深度交互分析质谱(deep interactome profiling by mass spectrometry,DIP-MS)” 的新方法,结合了AP和BN-PAGE分离(blue-native-PAGE separation)、数据非依赖性采集和深度学习信号处理。
3月26日,该研究以DIP-MS: ultra-deep interaction proteomics for the deconvolution of protein complexes为题发表在Nature Methods上。
研究人员将DIP-MS用于对蛋白质复合物进行深入的相互作用蛋白质组学分析,以解析复合物的组成。文章通过质谱技术和结构预测揭示了细胞中新的蛋白折叠底物与PAQosome和PFD(prefoldin)复合物的相互作用网络,拓展了对细胞蛋白质折叠调控机制的认识。
https://www.nature.com/articles/s41592-024-02211-y?utm_medium=external_display&utm_source=stork&utm_content=email&utm_term=null&utm_campaign=CONR_JRNLS_AWA1_CN_CNPL_0034V_STKRE
4.(J Hepatol,IF: 25.7)Review: 基于组学的脂肪肝疾病生物标记物的发现机遇与障碍

脂肪肝疾病患者迫切需要准确的生物标志物,以对纤维化和炎症进行分期和分级、监测疾病进展以及改善药物开发和审批流程。
高通量组学技术的快速发展和成本下降,再加上出色的计算能力,为开发新型生物标志物创造了黄金机遇,这些生物标志物能反映生物疾病过程,并能与多重分子系统相结合。因此,多组学可能会促进准确的个性化诊断时代的到来。
通过宿主遗传学、转录组学、蛋白质组学、代谢组学和脂质组学与肠道微生物、病毒和真菌宏基因组学和宏转录组学之间的相互作用,可以区分脂肪性肝病发生和发展的异质性。
文中提到,基于质谱的蛋白质组学的最新技术进步(包括样品制备自动化、液相色谱法的改进以及新型 质谱采集方法和复杂信息学解决方案的开发),使得研究人员可以在单一临床研究中生成数千份蛋白质组图谱。与此同时,研究人员已开始应用基于机器学习的分类算法来证明所提出的生物标记物对肝病的预测或鉴别能力。
无假设方法(Hypothesis-free approaches)揭示了组学技术在发现肝病生物标志物方面的潜力,并提出了比传统假设驱动研究更多的候选生物标志物。然而,这些基于全局组学的候选生物标志物很少经过独立队列的严格测试,也没有一个被应用于临床实践。
作者在文章结论中指出,未来五到十年,多组学的证据基础和成熟度将得到关键性的改善,从而使首批基于组学的生物标记进入临床实践,成为个性化医疗的精准工具。
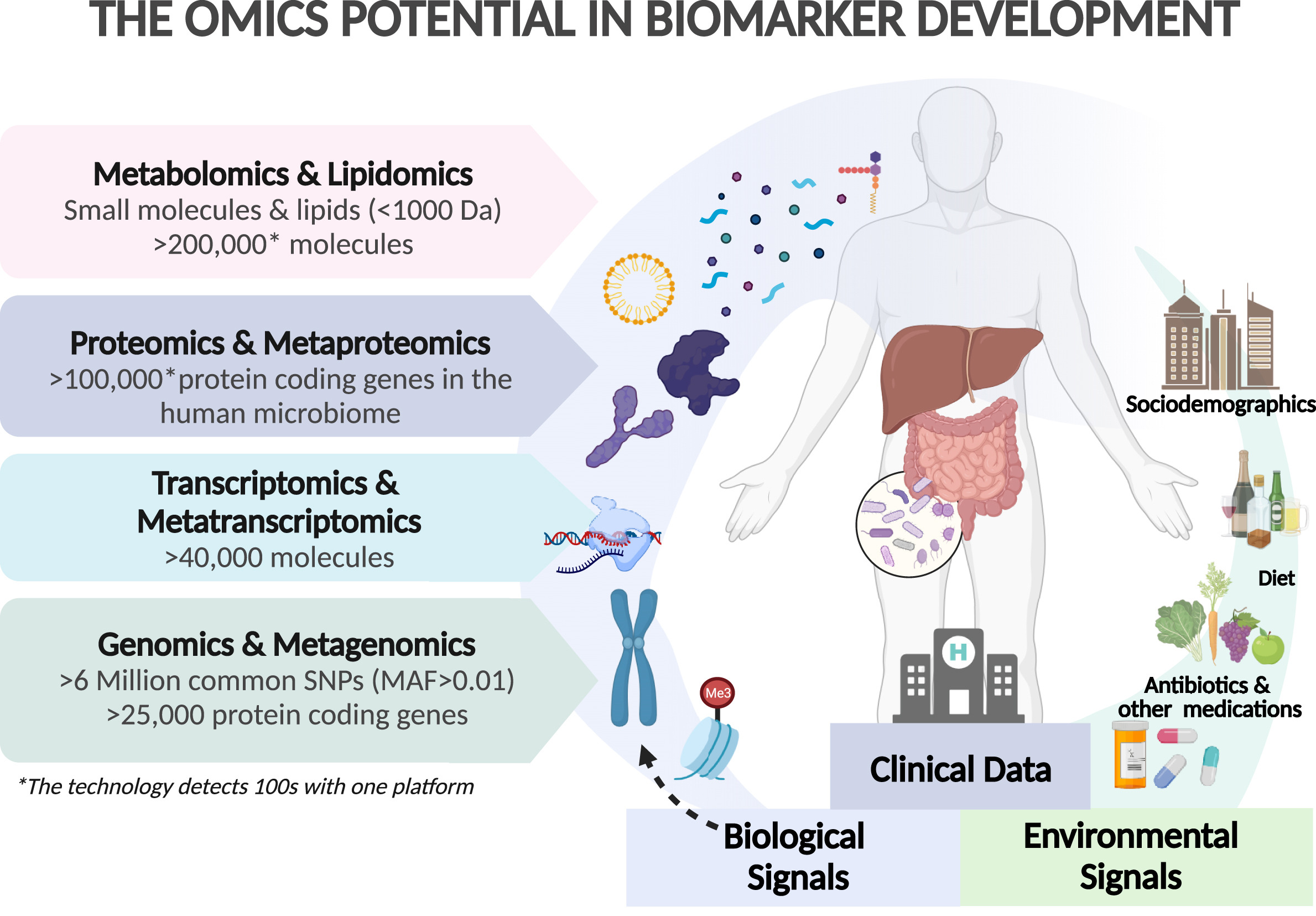
基于组学的生物标志物的潜力
https://www.journal-of-hepatology.eu/article/S0168-8278(24)00219-8/fulltext
5.(Nat Chem,IF: 21.8)内源性相分离蛋白质鉴定新策略

哺乳动物细胞内的相分离(phase separation)调节着与基因表达、信号传导、发育和疾病有关的生物分子凝聚物的形成。然而,大量的内源性凝聚物及其候选相分离蛋白尚未以定量和高通量的方式被发现。
本月,来自华中科技大学的研究人员提出了一种内源性相分离蛋白质鉴定的新策略。研究人员证明了通过对不同寡聚状态的蛋白质进行分类,可以在细胞蛋白质组中鉴定出内源性表达的生物分子凝聚体。他们利用体积压缩来调节细胞内蛋白质的浓度和拥挤程度,这是细胞生物分子凝聚体的物理调节因素。
蛋白质在凝结物中的分区或相分离程度的变化会导致蛋白质的寡聚状态发生变化,这些变化可通过密度梯度超速离心和定量质谱联用进行检测。研究人员总共鉴定了1518种内源性凝聚蛋白(condensate proteins),其中538种以前从未报道过。此外,他们还证明了其策略可以鉴定出响应特定生物过程的凝聚蛋白。
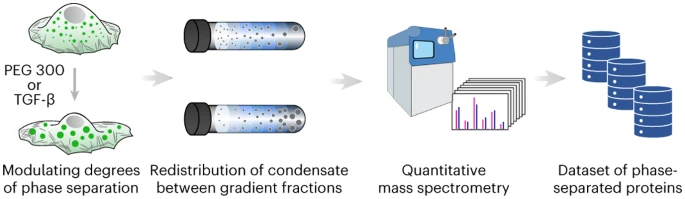
3月22日,研究人员就此策略在 Nature Chemistry 发表了 Research Briefing,文中提到:“与现有的公共数据库相比,我们的策略生成了更大的内源性生物分子凝聚物和相分离蛋白质的数据集,这一发展可能会将该领域带入定量和内源蛋白质组学新时代。”
https://www.nature.com/articles/s41557-024-01485-1?utm_medium=external_display&utm_source=stork&utm_content=email&utm_term=null&utm_campaign=CONR_JRNLS_AWA1_CN_CNPL_0034V_STKRE
简报链接:https://www.nature.com/articles/s41557-024-01503-2
6.(Nat Commun,IF: 16.6)肾脏多组学的基因推断揭示新的血压和高血压靶点

目前,血压(BP)调节的遗传机制仍不明确。利用肾脏特异性表观基因组注释和三维基因组信息,研究人员生成并验证了基因表达预测模型,以便在700个人类肾脏中进行全转录组关联研究。
研究人员确定了889个与血压相关的肾脏基因,其中399个被优先考虑为血压调节的贡献者。肾脏蛋白质组和microRNA组的推算发现了与血压相关的97种肾脏蛋白质和11种miRNA。与血浆蛋白质组学和代谢组学的整合揭示了肌醇、4-胍基丁酸酯和血管紧张素原的循环水平,它们是几个肾脏血压基因(SLC5A11、AGMAT、AGT)的下游效应因子。
该研究表明,由基因决定的肾脏表达量减少可能会模拟肾脏mRNA/蛋白质上罕见的功能缺失变体的效应,并导致血压升高(如ENPEP)。研究人员证明了从尿液和肾脏中提取的细胞之间蛋白质编码基因的表达具有很强的相关性(r = 0.81),这凸显了尿液细胞转录组学的诊断潜力。
总之,该研究为血压的遗传调控提供了新的生物学见解,有望推动高血压相关研究的临床转化。
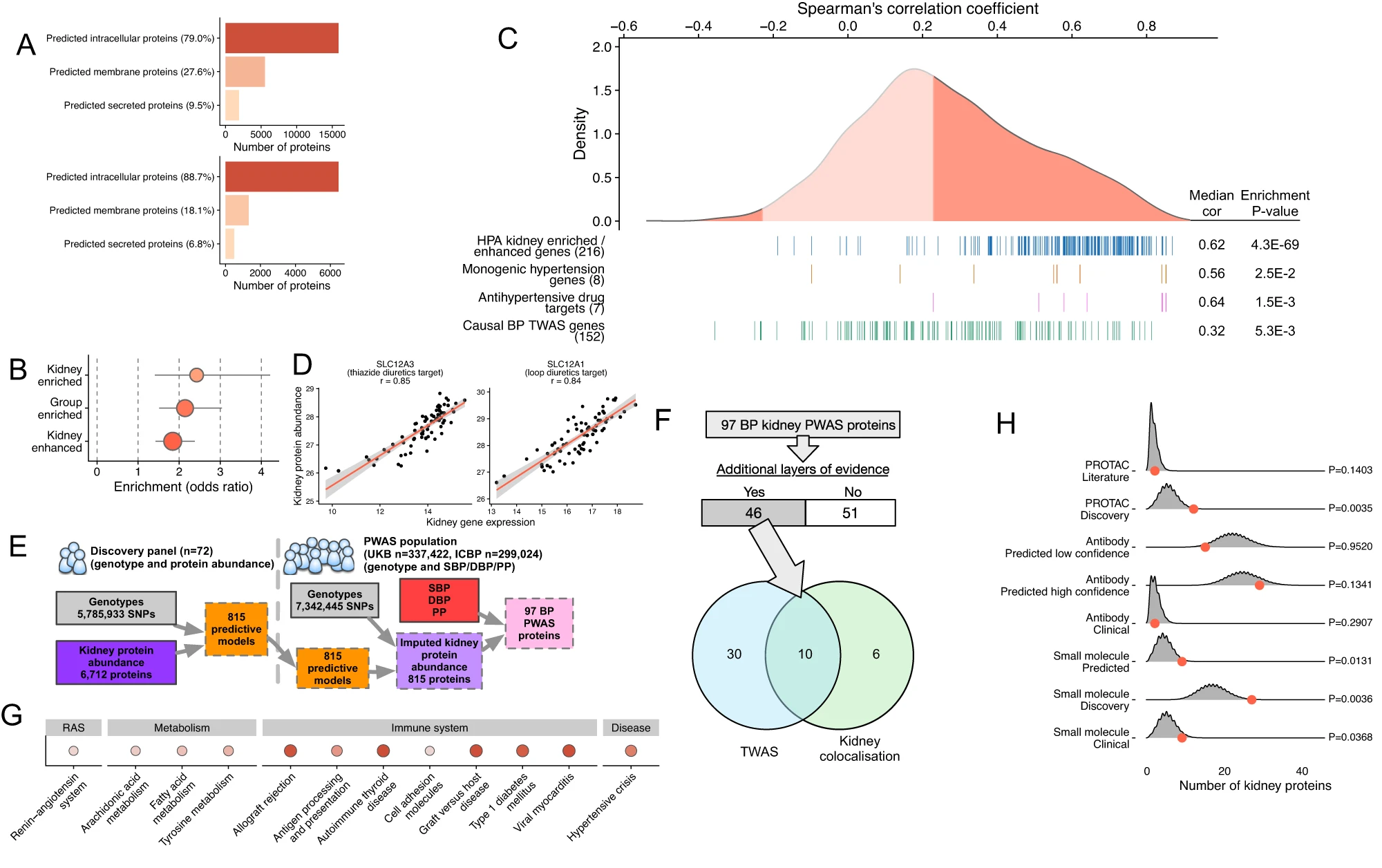
肾组织蛋白质组学与血压全蛋白质组相关联分析(PWAS)
https://www.nature.com/articles/s41467-024-46132-y?utm_medium=external_display&utm_source=stork&utm_content=email&utm_term=null&utm_campaign=CONR_JRNLS_AWA1_CN_CNPL_0034V_STKRE
7.(Nat Commun,IF: 16.6)通过深度学习预测糖肽片段质谱

近年来,深度学习在基于质谱的蛋白质组学中取得了显著的成功,目前正在糖蛋白质组学(glycoproteomics)领域崭露头角。虽然各种深度学习模型都能准确预测肽的片段质谱,但它们无法应对完整糖肽中的非线性聚糖结构(non-linear glycan structure)。
本月,浙江大学方群教授团队提出了一种基于深度学习的完整糖肽片段质谱预测方法DeepGlyco。该模型采用树状结构的长短期记忆网络来处理聚糖分子,并采用图神经网络架构来纳入特定聚糖结构的潜在片段路径。这一特征有利于模型对聚糖结构异构体的可解释性和区分能力。
研究人员进一步证明,预测的谱库可用于DIA糖蛋白组学,并期望这项工作能为糖蛋白组学提供有价值的深度学习资源。
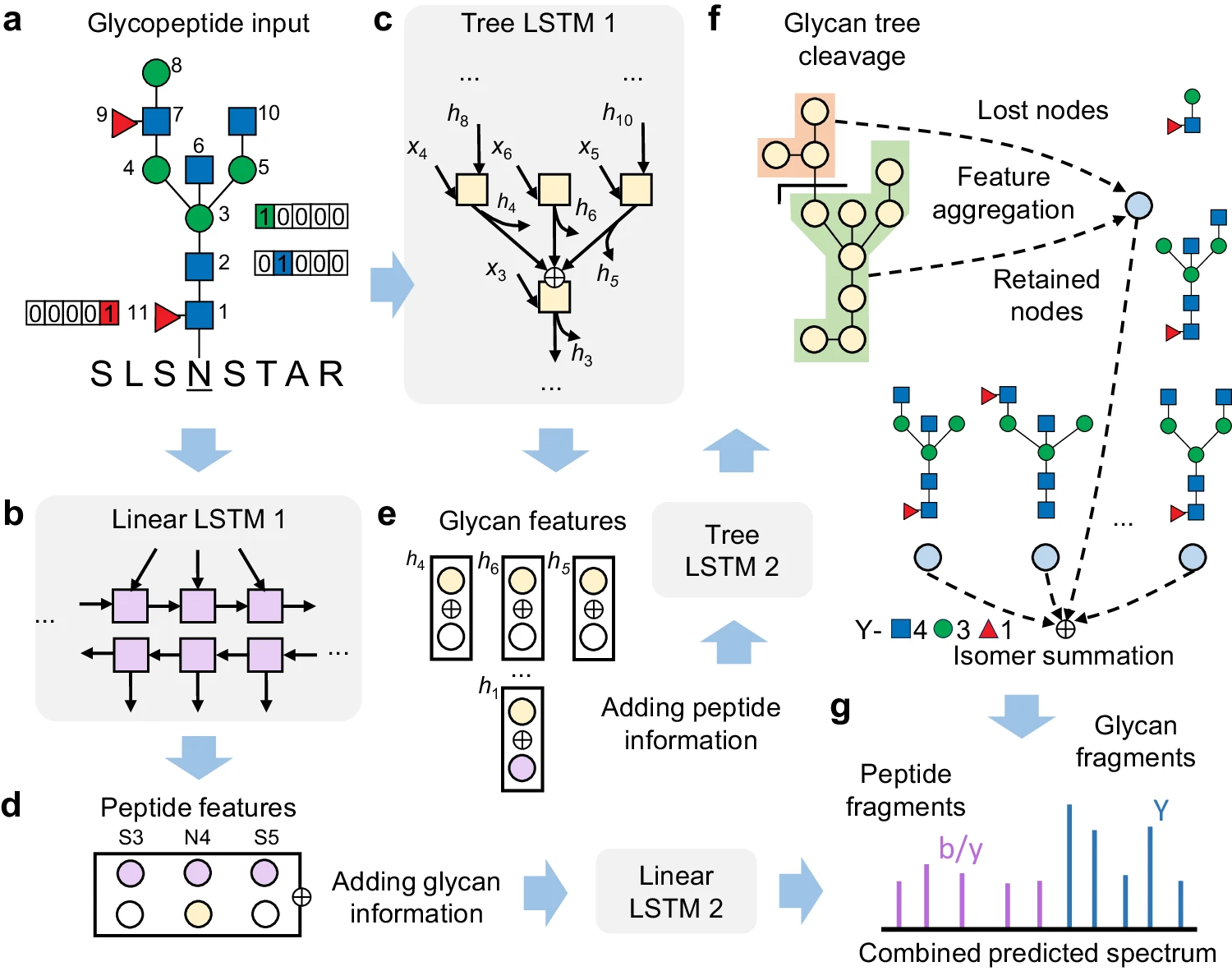
糖肽片段谱预测深度学习模型概述
https://www.nature.com/articles/s41467-024-46771-1?utm_medium=external_display&utm_source=stork&utm_content=email&utm_term=null&utm_campaign=CONR_JRNLS_AWA1_CN_CNPL_0034V_STKRE
8.(Nat Commun,IF: 16.6)One-Tip:全面覆盖最小细胞和单个胚胎的蛋白组学新方法

基于质谱(MS)的蛋白质组学工作流程通常涉及复杂的多步骤过程,或带来样品损失、可重复性差等挑战,需要大量的时间和资金投入以及专业技能。
在此,研究人员介绍了一种新的蛋白质组学方法One-Tip,它将高效的一锅样品制备(one-pot sample preparation)与精确的窄窗口式数据非依赖性采集(narrow-window data-independent acquisition,nDIA)分析无缝整合在一起。
One-Tip大大简化了样品处理过程,可从约1000 个HeLa细胞中重复鉴定出超9000种蛋白质。小鼠早期胚胎单细胞中约6000个蛋白质的nDIA鉴定凸显了One-Tip的多功能性。
此外,该研究还结合了Uno Single Cell Dispenser™,证明了One-Tip在单细胞蛋白质组学中的能力,每个HeLa细胞可鉴定出超过3000个蛋白质。
研究人员还将One-Tip工作流程扩展到分析从血浆中提取的细胞外囊泡(EV),从 16纳克EV制备物中鉴定出超过3000个蛋白质,证明了One-Tip的高灵敏度。One-Tip拓展了蛋白质组学的功能,为各种类型的样品提供了更高的深度和通量。
中国医学科学院苏州系统医学研究所叶子璐研究员为该研究的主要参与者。
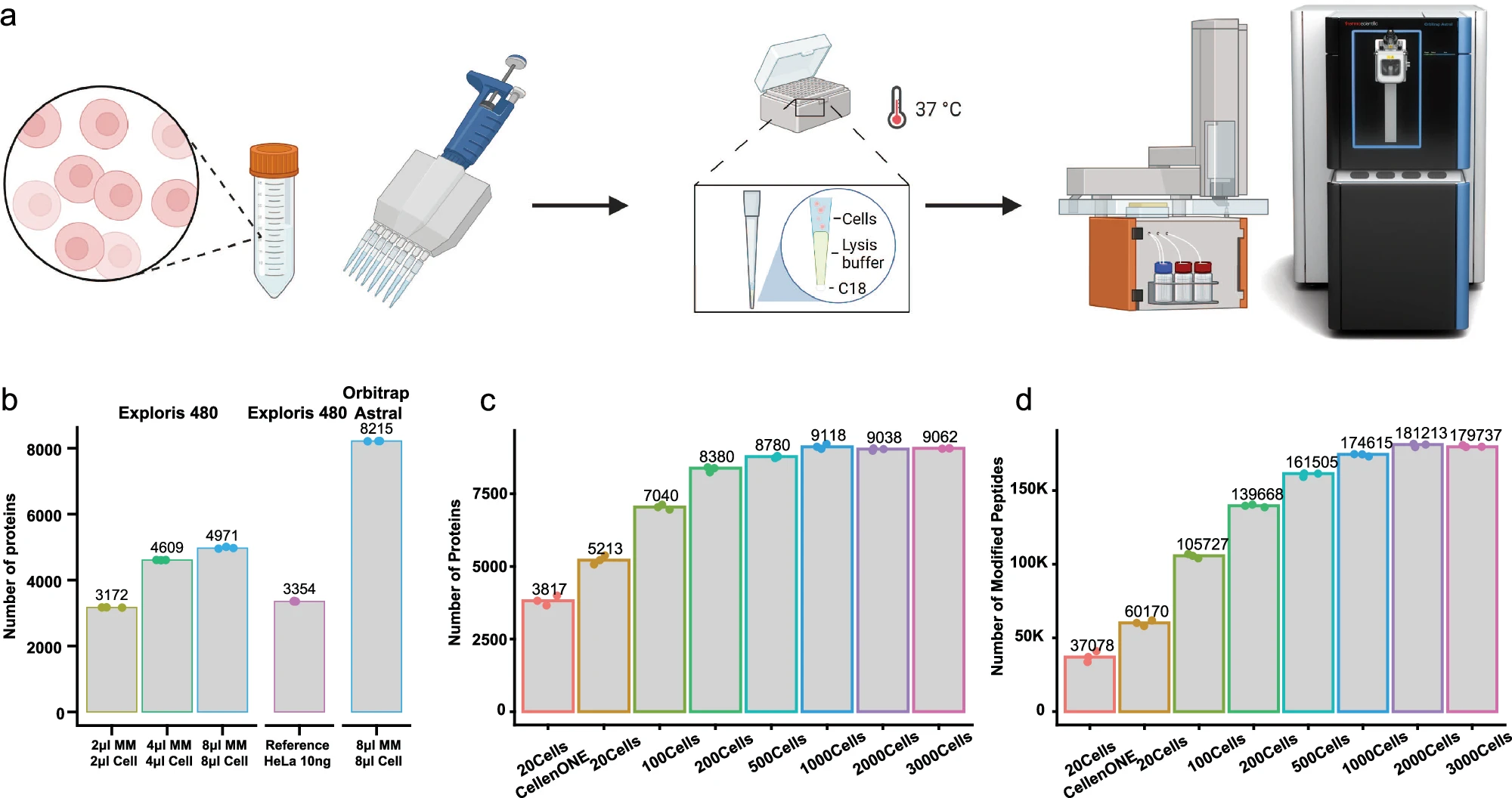
One-tip在单次分析中实现了近乎完整的蛋白质组深度
https://www.nature.com/articles/s41467-024-46777-9?utm_medium=external_display&utm_source=stork&utm_content=email&utm_term=null&utm_campaign=CONR_JRNLS_AWA1_CN_CNPL_0034V_STKRE
9.(Microbiome,IF: 15.5)ConDiGA:在宏蛋白组学中准确构建蛋白质序列数据库

宏蛋白质组学(Metaproteomics)可以提供一种直接的方法来鉴定微生物群中的微生物蛋白质,以确定其组成和功能特征。然而,由于微生物群样本的极端复杂性和高度多样性,深入和准确的宏蛋白组研究仍然受到限制。
在此,研究人员提出了一种从宏基因组数据中对基因进行精确分类注释的方法,即等位基因定向注释法(contigs directed gene annotation,ConDiGA),并利用该方法建立了用于宏蛋白质组分析的蛋白质序列数据库。
研究人员将其pipeline(ConDiGA或MD3)与另外两种流行的annotation pipelines(MD1和MD2)进行了比较。在MD1中,直接根据细菌全基因组数据库注释基因;在MD2中,根据细菌全基因组数据库注释等位基因,并将等位基因的分类信息分配给基因;在MD3中,以等位基因注释结果中最可信的物种作为注释基因的参考。比较了BLAST、Kaiju和 Kraken2等注释工具。
基于一个由12个物种组成的合成微生物群落,结果发现,在从宏基因组数据构建蛋白质序列数据库方面,使用MD3 pipeline的Kaiju优于其他工具。在粪便样本及模拟微生物群落和粪便样本的silico mixed数据集上也观察到了类似的表现。
该研究可以解决当前宏基因组衍生的蛋白质序列数据库中分类注释的可靠性问题,并可以促进微生物组的深入宏蛋白质组分析。
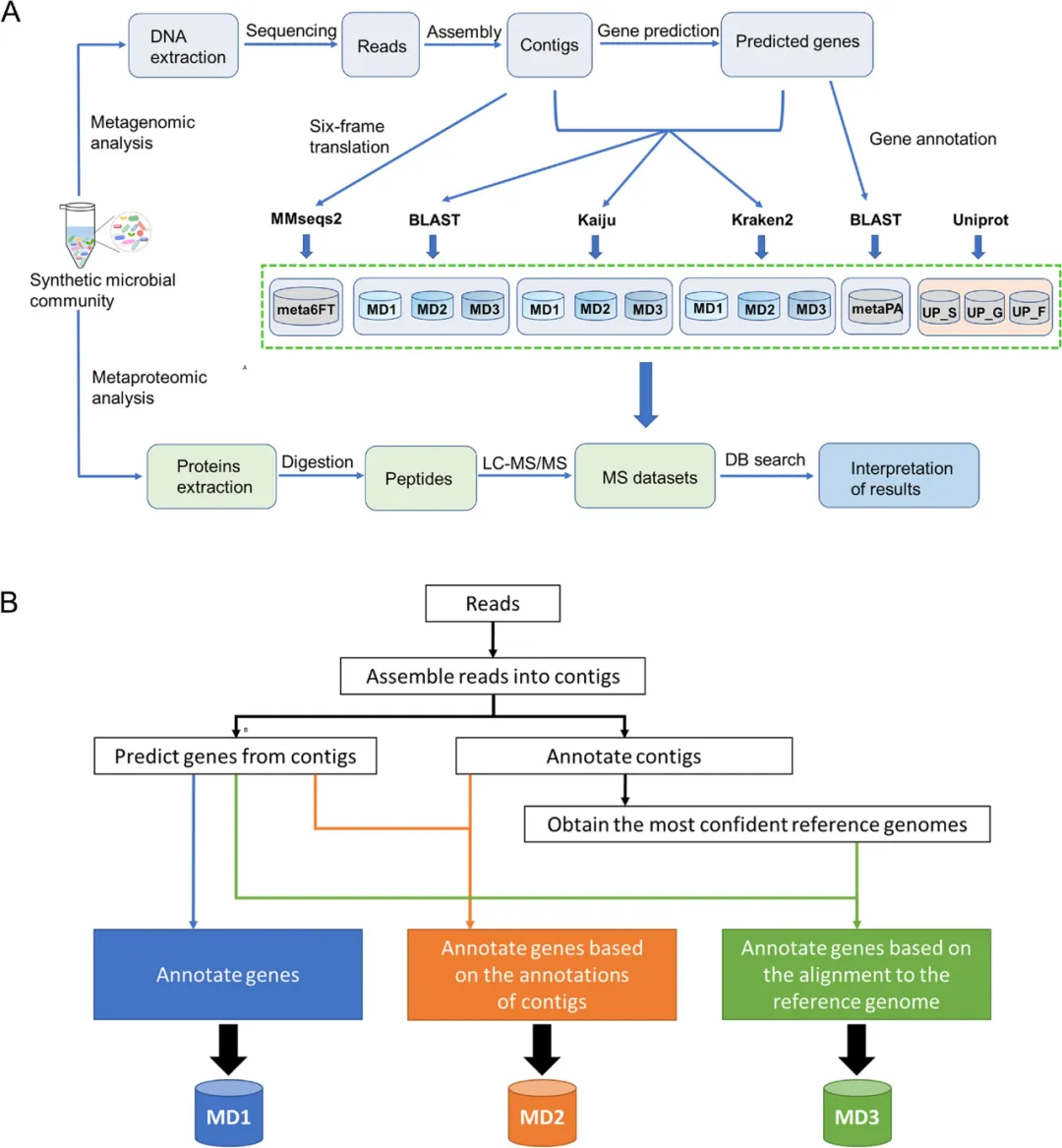
https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-024-01775-3?utm_medium=external_display&utm_source=stork&utm_content=email&utm_term=null&utm_campaign=CONR_JRNLS_AWA1_CN_CNPL_0034V_STKRE
10.(J Exp Med,IF: 15.3)TBK1 p.E696K突变导致小鼠渐冻症

TBK1基因的杂合子突变可导致肌萎缩侧索硬化症(ALS)和额颞痴呆(FTD)。大多数TBK1-ALS/FTD患者都携带有害的表达缺失突变,但目前尚不清楚是哪种TBK1功能导致了神经退行性变。
在此,研究人员探索了致病性TBK1错义变异p.E696K的影响,该变异并不影响蛋白质的表达,但会导致TBK1与自噬适配蛋白和TBK1底物optineurin结合的选择性缺失。通过细胞器特异性蛋白质组学研究(organelle-specific proteomics),研究人员发现在基因敲入小鼠模型和人类iPSC衍生的运动神经元中,p.E696K突变会导致神经元中自噬溶酶体功能障碍的症状提前出现,并引发受损溶酶体的积累。随之而来的是进行性、年龄依赖性运动神经元疾病。
与完全敲除Tbk1的小鼠的表型相反,没有发现依赖于RIPK/TNF-α的肝脏、神经元坏死和明显的自身炎症。体内研究结果表明,自噬溶酶体功能障碍是神经退行性变的一个触发因素,也是TBK1-ALS/FTD的一个有希望的治疗靶点。
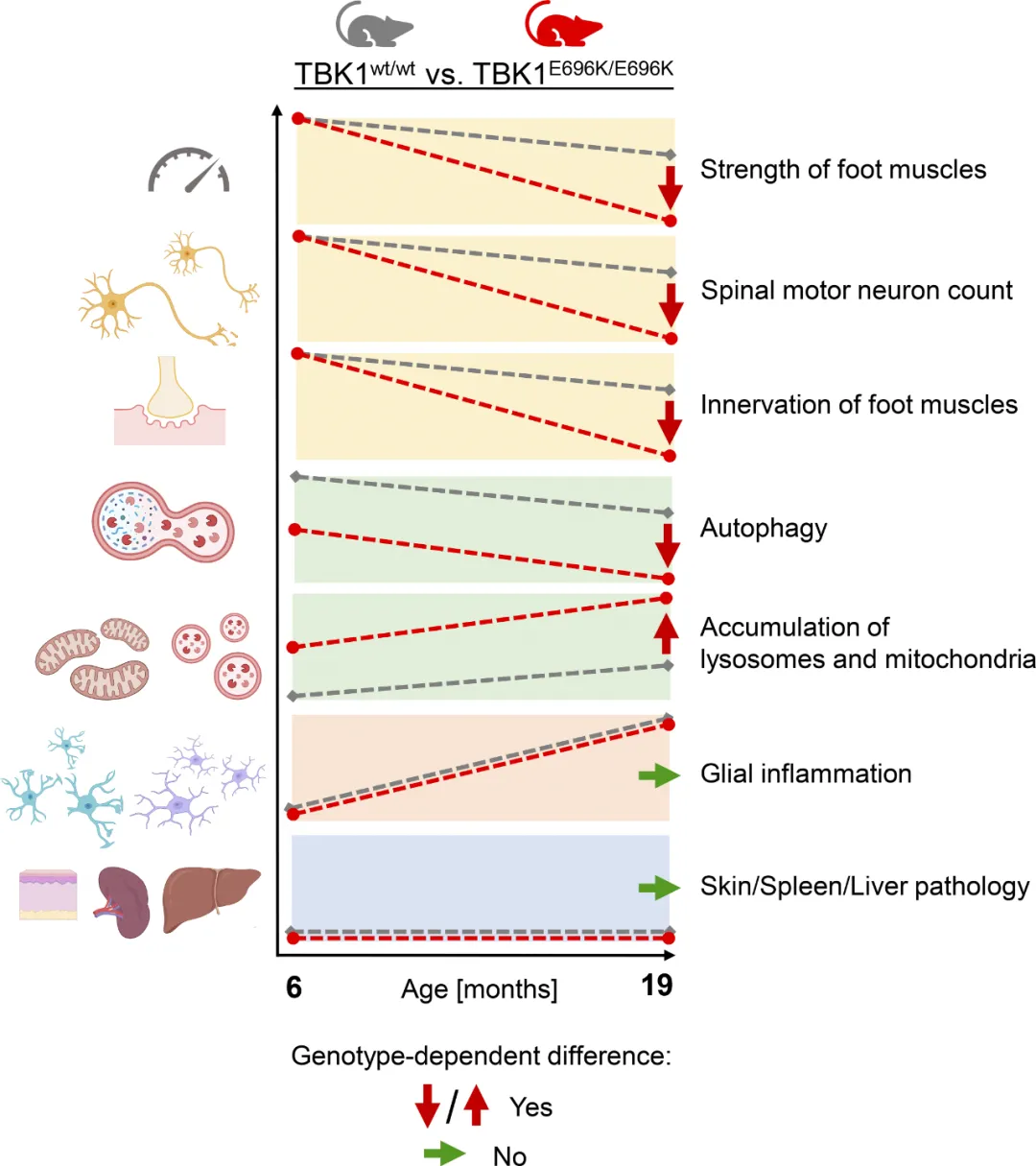
https://rupress.org/jem/article/221/5/e20221190/276649/A-TBK1-variant-causes-autophagolysosomal-and


